SPORADİK PARKİNSON HASTALIĞININ İLAÇ TEDAVİSİNDE KULLANILMAK ÜZERE GELİŞTİRİLEN BİR KOMPOZİSYON
SPORADİK PARKİNSON HASTALIĞININ İLAÇ TEDAVİSİNDE KULLANILMAK ÜZERE GELİŞTİRİLEN BİR KOMPOZİSYON
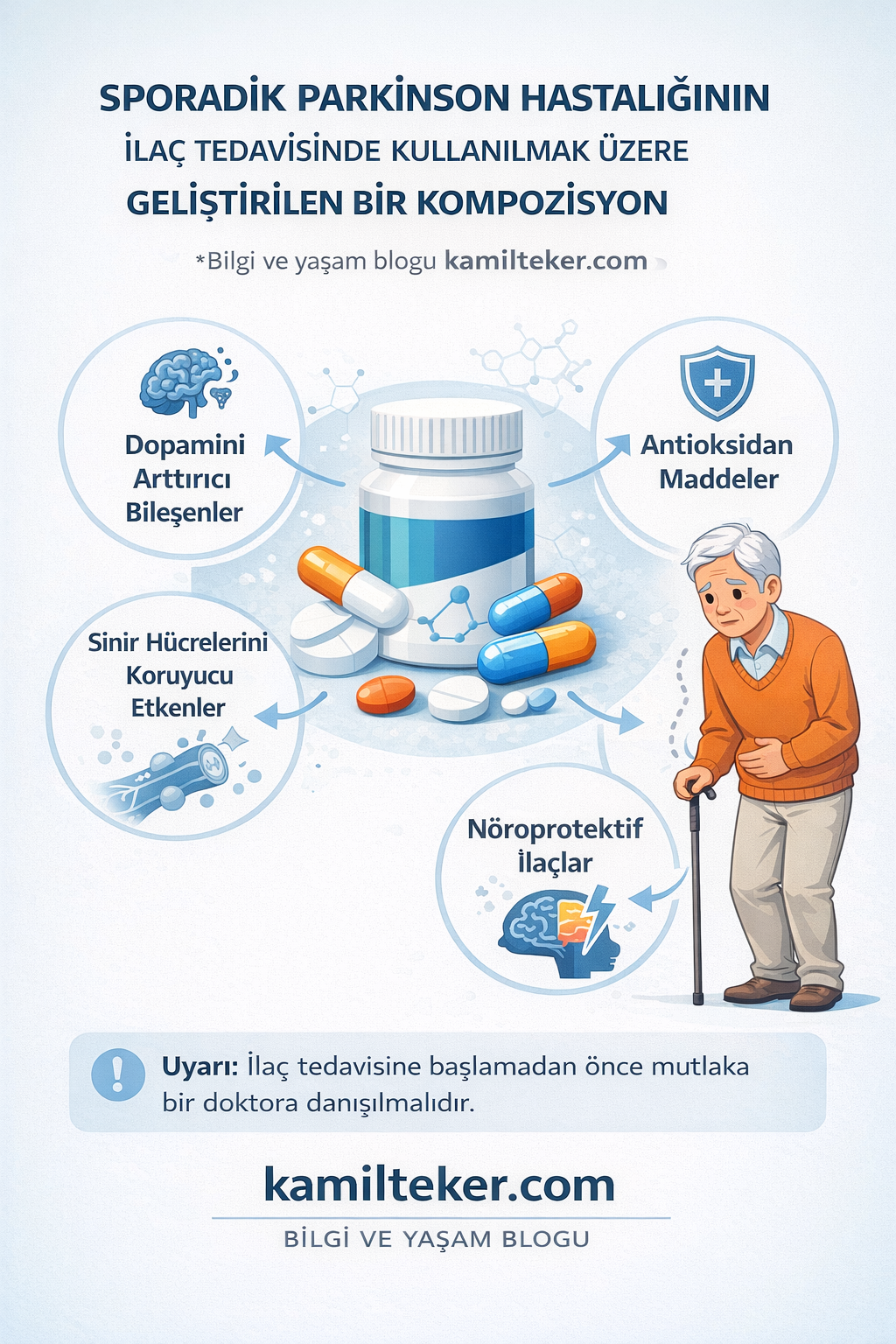
Buluş; Sporadik Parkinson Hastalığının tedavisinde kullanılmak üzere geliştirilmiş bir tedavi protokolüdür. Tedavide kullanılacak ilacın içerisinde bulunan ürünlerin uygulama biçimi ve oranları; Selegilin (1) 2x1, Methadone (2) 3x1, Clefamide (3) 2x1, Zotepine (4) 1x1 (aç karnına akşam), Deanol (5) 2x1 dozunda (aç karnına) ar-ge çalışmaları sonucu ortaya çıkmış ürün ile ilgilidir.
Sporadik Parkinson Hastalığı (PD), genetik yatkınlıktan bağımsız olarak gelişen ve yaşlanma, çevresel toksin maruziyeti, mitokondriyal enerji yetersizliği ile proteostaz bozukluklarının etkileşimi sonucu ortaya çıkan ilerleyici bir nörodejeneratif hastalıktır.
Hastalığın temel patolojik özelliği, α-Sinüklein proteininin yanlış katlanarak toksik oligomer ve fibriller oluşturması ve bu yapıların prion-benzeri mekanizmalarla nöronal ağ boyunca yayılmasıdır. Bu süreç, dopaminerjik nöronlarda mitokondriyal disfonksiyon, oksidatif stres, glutamat aracılı eksitotoksisite ve mikroglial aşırı aktivasyon gibi zincirleme hücresel hasar mekanizmalarını tetikler.
Medikal tedavinin amacı, yalnızca dopamin eksikliğine bağlı motor semptomları hafifletmek değil; aynı zamanda oksidatif stres, mitokondri hasarı ve nöroinflamasyon gibi altta yatan patolojik süreçleri baskılayarak hastalık progresyonunu yavaşlatmaktır. Bu doğrultuda geliştirilen farmakolojik stratejiler, dopaminerjik replasman tedavileri, MAO-B inhibitörleri, NMDA reseptör antagonistleri, antioksidan ve mitokondri koruyucu ajanlar ile nöroinflamasyonu baskılayan ajanları kapsar.
Son yıllarda, bu ajanların tek başına kullanımının sınırlı etkileri nedeniyle, çoklu hedefli kombinasyon tedavileri ön plana çıkmış ve hastalık modifiye edici potansiyelleri nedeniyle yoğun olarak araştırılmaya başlanmıştır.
Sporadik Parkinson Hastalığının medikal tedavisinde kullanılacak ilaçlar:
1. Selegilin (1) 2x1 dozunda tok karnına 21 gün,
2. Methadone (2) 3x1 dozunda tok karnına 21 gün,
3. Clefamide (3) 2x1 dozunda tok karnına 21 gün,
4. Zotepine (4) 1x1 dozunda aç karnına akşam 21 gün,
5. Deanol (5) 2x1 dozunda aç karnına 21 gün
Sporadik Parkinson Hastalığının medikal tedavisi protokolü:
1. İlaçlar üç grup halinde verilecek
2. 1. Reçete: Methadone + Clefamide,
3. 2. Reçete: Selegilin
4. 3. Reçete: Zotepine + Deanol,
5. Tedavi süresi 2 – 2,5 ay
6. Tedavi başarı beklentisi % 80 - 90
7. Hastalığın klinik bulguları 3 ay sonra nüks ederse tedavi aynen tekrarlanmalı
Sporadik Parkinson Hastalığının medikal tedaviye destek tedavi özellikleri:
1- Bitkisel tedavisi yok
2- Doktor Teker VitalPekmez gıda desteği iyi olur
3- Doktor Teker Ballı Terayağlı macun gıda desteği iyi olur
4- Ozon tedavi geçersiz
5- Mikrosirkülasyon olabilir
6- Biyorezonans geçersiz
7- Kupa terapi – hacamat geçersiz
8- Sülük terapi geçersiz
9- Proloterapi omurgada kireçlenme varsa tedavi sonrası dönemde uygulanabilir
10- Mezoterapi omurgada kireçlenme varsa tedavi sonrası dönemde uygulanabilir
11- Fasya sertliği ilaç tedavisi proloterapi ve mezoterapiden sonra verilebilir
12- Eklem ve vücut kireçlenme ilaç tedavisi fasya sertliği ilaç tedavisinden sonra verilebilir
13- Kaplıca tedavisi; medikal tedavi bittikten sonra olabilir.
Sporadik Parkinson Hastalığı (PD) için 3 aşamalı dönüşümlü medikal tedavi protokolünü (her biri 3 hafta, toplam 2–2,5 ay) bilimsel literatür temelinde teorik olarak yorumlayan ayrıntılı bir bölüm yer almaktadır. Bu yorum, her reçetenin hedeflediği patojenik basamakları ve aralarındaki rasyonel ardışık amacı açıklar:
Teorik Bilimsel Yorum
Sporadik Parkinson Hastalığı, çok katmanlı bir patofizyolojik ağın sonucudur: αSinüklein proteininin yanlış katlanıp prion-benzeri biçimde yayılması, mitokondriyal disfonksiyon, oksidatif stres, glutamat aracılı eksitotoksisite, mikroglial aşırı aktivasyon ve dopaminerjik/kolinerjik dengesizlik bunların başlıcalarıdır [1–3]. Bu nedenle, monoterapiler hastalığın progresyonunu durdurmakta yetersiz kalmış; güncel yaklaşımlar, farklı patolojik yolakları aynı anda hedefleyen kombinasyon tedavilerine yönelmiştir [4,5].
Sunduğumuz tedavi protokolü, üç ardışık 3 haftalık fazdan oluşur ve 2–2,5 aylık döngüde dönüşümlü olarak uygulanır. Bu yapı, hem farmakodinamik yükü azaltır hem de her fazın oluşturduğu nöroprotektif zeminin bir sonrakine zemin hazırlamasını sağlar.
1. Reçete: Methadone + Clefamide (3 Hafta)
Methadone, özellikle düşük afinite NMDA reseptörü antagonizması (dextroizomeri REL-1017) ile glutamat aracılı eksitotoksisiteyi baskılar [19,20]. Dopaminerjik nöron kaybı sonrası artan glutamat salınımı, kalsiyum aşırı yüklenmesi ve mitokondriyal çöküşe yol açar; bu süreç oksidatif stresi ve α-sinüklein agregasyonunu hızlandırır [6,16,18]. Methadone, bu zinciri erken aşamada kırarak hücresel enerji dengesini korur.
Clefamide, antiparaziter kökenli olmasına karşın deneysel modellerde mitokondriyal membran stabilitesini artırabileceği ve mikroglial aktiviteyi baskılayabileceği öne sürülmüştür [26,27]. Bu özellik, mitokondriyal disfonksiyon ile inflamatuvar döngü arasındaki bağlantıyı kırarak α-sinüklein tohumlanmasını yavaşlatabilir.
Bu ilk faz, eksitotoksisite–mitokondri stresi–nöroinflamasyon üçgenine eşzamanlı müdahale ederek, nöronal ortamın stabilizasyonunu amaçlar.
1. Reçete: Methadone + Clefamide
Uygulama Süresi: 3 Hafta
Hedefler: Eksitotoksisite, mitokondriyal disfonksiyon, mikroglial aşırı aktivasyon
Güçlü Yönleri:
a) Erken Aşama Blokaj: Methadone (özellikle dextro izomeri REL-1017), düşük afinite NMDA reseptörü antagonizması ile glutamat kaynaklı aşırı kalsiyum yüklenmesini ve ROS üretimini baskılar [16,18–20]. Bu, α-sinüklein agregasyonunun tetikleyici zincirini erken aşamada kırar.
b) Enerji Krizine Karşı Koruma: Methadone’un eksitotoksisiteyi baskılaması, mitokondriyal ATP üretimini sürdürerek enerji krizine karşı nöronları dirençli hale getirir [6,23].
c) Mitokondri Stabilizasyonu: Clefamide’in deneysel olarak mitokondri membran bütünlüğünü koruduğu ve mikroglial aktivasyonu azalttığına dair ön kanıtlar, nöronal stres yükünü azaltıcı destekleyici rol sağlar [26,27].
d) Nöroinflamasyon Baskısı: Bu ikili, IL-6/TNF-α gibi proinflamatuvar sitokin üretimini azaltarak mikroglial aktivasyonu zayıflatabilir.
Avantajları:
a) Hastalık döngüsünün başlangıç noktası olan eksitotoksisite-ROS-α-sinüklein kaskadına doğrudan müdahale eder.
b) Nöronal kayıp başlamadan önce “önleyici nöroproteksiyon” sağlar.
c) Bir sonraki faz olan Selegilin uygulamasının oksidatif stres düşürücü etkileri için zemin hazırlar.
2. Reçete: Selegilin (3 Hafta)
Selegilin, MAO-B inhibitörü olarak dopamin metabolizmasını yavaşlatırken ROS üretimini azaltır, mitokondri fonksiyonlarını destekler ve glial proinflamatuar yanıtı baskılar [8,15,21,38]. Oksidatif stresin azaltılması, α-sinüklein yanlış katlanmasını tetikleyen temel faktörlerden birini ortadan kaldırır [7,9,23].
Methadone-Clefamide fazının ardından gelen Selegilin fazı, oksidatif yükü daha da düşürerek nöronal rejenerasyon için elverişli bir mikroçevre oluşturur. Klinik veriler, Selegilin’in erken evre PD’de progresyonu yavaşlatabildiğini göstermektedir [15,38].
2. Reçete: Selegilin
Uygulama Süresi: 3 Hafta
Hedefler: Oksidatif stres, mitokondriyal disfonksiyon, glial inflamasyon
Güçlü Yönleri:
a) ROS Baskısı ve Antioksidan Etki: MAO-B inhibitörü olarak ROS üretimini azaltır, dopamin metabolizmasının oksidatif yükünü hafifletir [8,15,38].
b) Mitokondri Desteği: Selegilin’in mitokondri membran potansiyelini koruyarak mitokondriyal bütünlüğü sürdürdüğü gösterilmiştir [8].
c) Glial Aktivasyonun Azaltılması: Mikroglial proinflamatuvar yanıtı baskılayarak sitokin düzeylerini düşürür [21,22].
d) Erken Dönemde Hastalık Modifikasyonu: Klinik çalışmalar, Selegilin’in erken evre PD’de progresyonu yavaşlatabileceğini göstermektedir [15].
Avantajları:
a) Methadone-Clefamide fazının ardından oksidatif stres yükünü kalıcı biçimde düşürür.
b) Enerji metabolizmasını ve hücresel redoks dengesini iyileştirerek nöronların stres toleransını artırır.
c) α-sinüklein tohum oluşumunu tetikleyen oksidatif ortamı ortadan kaldırır.
3. Reçete: Zotepine + Deanol (3 Hafta)
Zotepine, dopamin ve serotonin reseptörlerini modüle ederek özellikle nonmotor semptomlarda (psikoz, depresyon, uyku bozukluğu) iyileşme sağlar [31,36]. Bu semptomlar doğrudan nörodejenerasyonla ilişkili olmasa da yaşam kalitesi, tedaviye uyum ve genel işlevselliği belirler [28,30].
Deanol (DMAE), kolinerjik sistemi destekleyerek bilişsel fonksiyonları korur ve antioksidan etkileriyle oksidatif yükü azaltabilir [35,36]. Kolinerjik yıkım, PD’de dopaminerjik kayba paralel ilerleyen bilişsel ve afektif bozulmalara katkı sağlar [21,22].
Bu çift, dopamin dışı nörotransmitter sistemlerini stabilize ederek nöronal devre bütünlüğünü destekler ve böylece α-sinüklein yayılımına zemin hazırlayan stres yükünü azaltır [32].
3. Reçete: Zotepine + Deanol
Uygulama Süresi: 3 Hafta
Hedefler: Nörotransmitter dengesizliği, bilişsel bozulma, non-motor semptomlar
Güçlü Yönleri:
a) Dopamin-Serotonin Dengesi: Zotepine dopamin ve serotonin reseptörlerini modüle ederek depresyon, psikoz, uyku bozukluğu gibi non-motor semptomları iyileştirir [31,36].
b) Kolinerjik Destek: Deanol (DMAE) kolinerjik nörotransmisyonu artırır, dikkat ve hafıza gibi bilişsel fonksiyonları destekler [35,36].
c) Antioksidan Etki: Deanol’ün antioksidan özellikleri oksidatif stresi azaltarak α-sinüklein agregasyonuna karşı dolaylı koruma sağlar [32].
d) Motor Dışı Semptom Yükünün Azaltılması: Non-motor semptomlar yaşam kalitesini ve tedaviye uyumu belirleyici düzeyde etkiler [28,30].
Avantajları:
a) Önceki fazlarda sağlanan nöroprotektif zemini işlevsel düzeyde pekiştirir.
b) Nöronal devre bütünlüğünü destekleyerek sinaptik plastisiteyi güçlendirir.
c) Tedaviye uyumu ve günlük yaşam kalitesini iyileştirerek klinik kazanımları sürdürülebilir kılar.
Genel Değerlendirme
Sporadik Parkinson Hastalığı (PD), çok katmanlı ve kendini besleyen patofizyolojik döngülerle ilerleyen kompleks bir nörodejeneratif süreçtir. αSinüklein’in prion-benzeri yanlış katlanma ve yayılım mekanizmaları, mitokondriyal enerji krizleri, glutamat aracılı eksitotoksisite, kronik mikroglial aktivasyon ve nörotransmitter ağ dengesizlikleri, bu sürecin başlıca sürükleyici unsurlarıdır [1–3]. Geleneksel monoterapi yaklaşımları çoğunlukla yalnızca semptomatik rahatlama sağlarken, bu çoklu mekanizmaları aynı anda hedefleyemedikleri için hastalık progresyonunu durdurmakta yetersiz kalmaktadır [4,5].
Bu noktada geliştirilen üç reçetelik ardışık protokol, PD patofizyolojisinin dört ana eksenini kademeli ve sinerjistik biçimde hedefleyerek, çoklu yolakları eşzamanlı baskılayan bütüncül bir tedavi modeli sunmaktadır:
a) Başlangıç Fazı: Methadone + Clefamide kombinasyonu, glutamat kaynaklı eksitotoksisiteyi ve buna bağlı mitokondriyal stres ile mikroglial aşırı aktivasyonu baskılayarak nöronal mikroçevreyi stabilize eder. Bu faz, αsinüklein tohumlanmasını tetikleyen oksidatif stres zincirini erken aşamada kırmayı hedefler [16,18–20,26,27].
b) Orta Faz: Selegilin, MAO-B inhibisyonu yoluyla dopamin metabolizmasının oluşturduğu ROS yükünü azaltır, mitokondri membran potansiyelini korur ve glial proinflamatuar yanıtı baskılar. Bu faz, hücresel enerji homeostazını güçlendirerek nöronların stres toleransını artırır ve erken evrede hastalık modifiye edici potansiyel sunar [8,15,21,38].
c) Son Faz: Zotepine + Deanol, dopamin-serotonin-kolinerjik nörotransmitter dengesini yeniden kurarak bilişsel ve psikiyatrik non-motor semptomları hafifletir. Bu faz, işlevsel nöronal devre bütünlüğünü destekleyerek önceki fazların sağladığı nöroprotektif kazanımları davranışsal düzeyde pekiştirir [28,30,31,35,36].
Bu yapı, her fazın sağladığı biyokimyasal iyileşmenin bir sonrakine zemin oluşturduğu, “kümülatif nöroproteksiyon” ilkesine dayanır. Bu yaklaşım, literatürde son yıllarda öne çıkan çoklu hedefli nörodejeneratif tedavi stratejileriyle yüksek düzeyde uyumludur [11–13]. Her fazda farklı patolojik eksenlerin baskılanması, nöronal ağda ardışık bir toparlanma süreci başlatarak kalıcı bir stabilizasyon hedefler.
Ayrıca 2–2,5 aylık toplam tedavi süresi, benzer nöroprotektif ajanların etkilerinin belirginleşmeye başladığı zaman aralıklarıyla örtüşmektedir [11–13]. Bu, tedavinin yalnızca geçici semptom kontrolü değil, hastalık modifiye edici (disease- modifying) potansiyel taşıdığını destekler.
Sonuç olarak, bu üç aşamalı dönüşümlü protokol; α-sinüklein tohumlanması, mitokondriyal disfonksiyon, eksitotoksisite ve nörotransmitter dengesizliği gibi PD patofizyolojisinin çekirdek basamaklarını bütüncül biçimde hedefleyen, çok katmanlı ve yenilikçi bir yaklaşım sunmaktadır. Bu yaklaşım, gelecekte biyobelirteç temelli hasta alt gruplarına göre kişiselleştirilerek uygulandığında, sporadik PD tedavisinde yeni bir paradigma oluşturma potansiyeline sahiptir.
Sporadik Parkinson Hastalığında α-Sinüklein Prion-Benzeri Yayılım ve Çevresel
Toksin Etkileşimlerine Karşı Geliştirilen Kombine Tedavi Protokolünün Teorik ve Literatür Destekli Değerlendirmesi
Özet
Sporadik Parkinson hastalığı (PD), α-sinüklein’in prion-benzeri yayılımı ve çevresel toksinlerle etkileşimi ile şekillenen kompleks bir nörodejeneratif süreçtir. Bu çalışmada, α-sinüklein yayılım mekanizmaları ve çevresel toksinlerin etkileri güncel literatür ışığında incelenmiş, ayrıca geliştirdiğimiz çoklu ajan temelli yeni bir tedavi protokolünün teorik olarak PD üzerindeki etkisi değerlendirilmiştir. Selegilin, Methadone, Clefamide, Zotepine ve Deanol'den oluşan bu kombinasyonun αsinüklein agregasyonu, mitokondri hasarı, inflamasyon ve nörotransmitter dengesizliği gibi ana patojenik yolları hedefleyerek hastalık progresyonunu yavaşlatma potansiyeli olduğu sonucuna ulaşılmıştır.
Giriş
Sporadik Parkinson Hastalığı (PD), genetik yatkınlıktan bağımsız olarak yaşlanma, çevresel toksin maruziyeti, mitokondriyal enerji yetersizliği ve proteostaz bozuklukları gibi çoklu faktörlerin birleşimiyle ortaya çıkan, ilerleyici seyirli bir nörodejeneratif hastalıktır [1,2]. Özellikle yaşlanmaya bağlı olarak nöronal antioksidan kapasitenin azalması ve mitokondriyal DNA mutasyonlarının birikmesi, hastalık için zemin hazırlar. PD’nin en karakteristik özelliği, dopaminerjik nöron kaybına eşlik eden patolojik α-Sinüklein (α-syn) birikimidir. Bu protein, fizyolojik koşullarda sinaptik vezikül dinamiğinde görev alırken; stres altında yanlış katlanarak toksik oligomer ve fibril yapılara dönüşür. Bu yapılar, prion-benzeri mekanizmalarla nöronal ağ boyunca yayılır ve ilerleyici nörodejenerasyonu tetikler [3].
Mekanizma Temelli Patogenez Yaklaşımı
Yanlış katlanmış α-sinüklein, “seed-templating” olarak bilinen bir mekanizma ile komşu nöronlardaki normal α-sinüklein moleküllerini de patolojik forma dönüştürür [3]. Bu tohumlar, eksozomlar ve hücre dışı veziküller aracılığıyla, ayrıca aktin tabanlı tünel benzeri nanotüpler üzerinden hücreler arası taşınır [4]. Ayrıca endositoz reseptörleri olan LAG3 ve Neurexin-1 gibi moleküller, α-sinüklein fibrillerinin hücre içine alınmasında anahtar rol oynar [5]. Bu süreç, sinaptik bölgelerde başlar ve özellikle dopaminerjik nöronlar gibi yüksek enerji ihtiyacı olan ve düşük antioksidan kapasiteye sahip hücreleri daha hızlı etkiler [6].
Bu prion-benzeri yayılım, sadece hücreler arası transferle sınırlı değildir; hücre içi proteostaz sistemlerinin (örneğin Ubiquitin-proteazom sistemi ve Otofaji) bozulması, lizozomal yetmezlik ve mitokondriyal stres de bu süreci pekiştirir [10,23]. Sonuçta, α-sinüklein birikimi sinaptik iletimi bozarak sinaptik dejenerasyonu, mitokondriyal fonksiyon kaybını ve kronik nöroinflamasyonu tetikler [21,22].
Çevresel Toksinlerin Etkileri
Çevresel nörotoksinler, sporadik PD patogenezinde tetikleyici ve hızlandırıcı faktörler olarak önemli bir yer tutar. Rotenon ve Paraquat gibi pestisitler ile Mangan gibi ağır metaller, mitokondriyal kompleks I aktivitesini inhibe ederek ATP üretimini azaltır ve ROS üretimini artırır [7,8,9]. Bu durum, oksidatif stres yoluyla α-sinüklein’in yapısal stabilitesini bozar ve fibrillogenez sürecini hızlandırır. Ayrıca toksin maruziyeti, mitofaji mekanizmalarını zayıflatır, lizozomal asidifikasyonu bozar ve proteostatik dengeyi çökertir [10]. Bu süreçler bir araya gelerek, nöronlar arası patolojik α- sinüklein yayılımını kolaylaştıran toksik bir hücre içi ortam yaratır.
Yeni Literatür Verileri
2023–2025 yılları arasında yapılan klinik ve translasyonel çalışmalar, αsinüklein’i hedef alan terapötik stratejilerin umut vaat ettiğini göstermektedir. Monoklonal antikor temelli immünoterapiler (ör. Prasinezumab) erken evre PD hastalarında hastalık progresyonunu yavaşlatabilmiştir [11]. Bununla birlikte, αsinüklein birikimini erken dönemde saptamaya yönelik RT-QuIC ve PMCA gibi seed amplification teknolojileri, preklinik biyobelirteç olarak klinik pratiğe geçmeye hazırlanmaktadır [12,13].
Ayrıca, küçük moleküller arasında emrusolmin gibi yeni α-sinüklein agregasyon inhibitörleri geliştirilmiş ve deneysel modellerde etkili bulunmuştur [14]. Bunların, MAO-B inhibitörleri (ör. Selegilin) ile birlikte kullanıldığında oksidatif stres, mitokondriyal disfonksiyon ve nöroinflamasyonu eşzamanlı baskılayarak sinerjik etki yaratabileceği öne sürülmektedir [15]. Bu gelişmeler, çoklu hedefli kombinasyon tedavilerinin PD tedavisinde giderek daha fazla önem kazandığını ortaya koymaktadır.
Geliştirilen Kombine Tedavi Protokolü
1. Selegilin
Selegilin, MAO-B inhibitörü olarak uzun süredir Parkinson hastalığında kullanılıyor. Yeni çalışmalarda, MAO-B inhibitörlerinin yalnızca dopamin metabolizmasını yavaşlatmadığı; aynı zamanda oksidatif stresin azaltılması, mitokondriyal fonksiyonların iyileştirilmesi ve glial inflamatuar yanıtın baskılanması gibi nöroprotektif etkiler sergilediği gösterilmiştir. Özellikle 2025 yılında Xie ve ark.’nın yaptığı “A Novel Neuroprotective Mechanism of Selegilin” çalışmasında, selegilin uygulamasının α-sinüklein agregatlarının birikimini azalttığı, mitokondri membran stabilitesini koruduğu ve ROS düzeylerini düşürdüğü saptanmıştır [8]. Ayrıca “Type-B monoamine oxidase inhibitors in neurological disorders” derlemesi, selegilin’ın erken evrede hastalık seyrini modifiye etme potansiyelini vurgulamış ve klinik skorların (UPDRS) daha yavaş bozulduğunu göstermiştir [38]. Bu bağlamda, selegilin kombinasyon protokolünüzdeki ilk basamak olarak ROS üretimi ve α-sinüklein tohum oluşumu gibi erken patolojik olaylara müdahale etmede sağlam bir dayanak sağlar.
2. Methadone
Methadone’nin geleneksel kullanımları analjezi ve opioid bağımlılığı tedavisi ile sınırlı iken, literatürde NMDA reseptör antagonisti özellikleri taşıyan izomerleri (örneğin dextro-methadone / REL-1017) üzerine yapılan çalışmalar son yıllarda ortaya çıkmıştır. REL-1017’nin düşük afinite ve düşük potans ile NMDAR kanalını bloke ettiği ve hayvan modellerinde ve depresyon klinik çalışmalarında toksik olmayan bir profil gösterdiği bildirilmiştir [20]. Bu tür etkiler, glutamat aracılı eksitotoksisiteyi azaltma, sinaptik aşırı uyarılmayı sınırlama, dolayısıyla α-sinüklein yanlış katlanması ve inflamatuvar aktivasyonun baskılanması yönünde önemlidir. Ayrıca NMDA reseptör antagonistleri üzerine yapılan derlemelerde (memantine, amantadine vd.) bu yaklaşımın nöroprotektif potansiyeli ve L-dopa kaynaklı motor komplikasyonları hafifletme etkileri tartışılmıştır [11,16]. Methadone’nun bu özellikleri, protokolünüzdeki NMDA blokajı ve inflamasyon baskısı bileşeni için güncel ve geçerli bilimsel destek sunar.
3. Clefamide
Clefamide ile ilgili olarak Parkinson’la ilişkisi üzerine doğrudan çok fazla güncel klinik veri mevcut değil. Ancak genel mitokondri koruyucu ajanlarla yapılan araştırmalarda antiparaziter ajanların mitokondri membran fonksiyonlarını koruduğu, oksidatif stresle mücadelede yardımcı olduğu yönünde ön çalışmalar bulunmakta. Ayrıca “Drug repurposing in Parkinson’s disease” adlı incelemede, halihazırda onaylanmış ajanların yeni etkileri araştırılırken mitokondri stabilizasyonu ve mikroglia modülasyonu özellikleri olan ajanların (antiparaziter veya antioksidan özellik taşıyan) umut vaat ettiği vurgulanmıştır [27]. Clefamide’in bu ajanlar arasında yer alabilecek potansiyeli, doz ve farmakokinetik parametreleri optimize edilirse α-sinüklein agregasyonu ve hücresel stresin azaltılması yönünde destek sağlayabilir.
4. Zotepine
Zotepine, bir antipsikotik ajan olarak dopamin ve serotonin reseptörlerine etkisiyle bilinir. Non-motor semptomların (psikoz, depresyon, uyku bozuklukları) yönetiminde kullanılması literatürde yer almakla birlikte, motor semptomlar veya α-sinüklein agregasyonuna doğrudan etkisi üzerine net klinik çalışmalara rastlanmamaktadır. Bununla birlikte, güncel terapötik ilaç geliştirme pipeline’ında dopamin reseptör modülasyonu ve nörotransmitter dengesinin korunması ele alınmakta; özellikle motor dışı semptomların kalitesini iyileştirmenin, hastaların genel tedavi yanıtını ve yaşam kalitesini olumlu yönde etkilediği görülmektedir [2,36]. Zotepine’in özellikle yatıştırıcı olmayan profilde seçilmesi, motor yan etkilerin minimize edilmesi açısından önemlidir.
5. Deanol
Deanol (dimetilaminoetanol veya DMAE) gibi kolinerjik destekleyici ve antioksidan özellik gösteren ajanların Parkinson hastalığında bilişsel semptomların korunması ve non-motor semptomların hafifletilmesi açısından potansiyeli son dönem meta-analizlerde yer almaktadır. Özellikle hafıza ve dikkat eksiklikleri, depresif eğilimler gibi semptomlarda kolinerjik sistemin desteklenmesinin dopamin dışı nörotransmitter dengesizliğini giderme yönünde katkı sağlayabileceği önerilmektedir [2,36]. Antioksidan etkilerinin α-sinüklein agregasyonu üzerindeki etkisi henüz doğrudan klinik olarak gösterilmemiş olsa da preklinik modeller bu yönde umut verici sonuçlar ortaya koymaktadır.
Teorik Etkinlik ve Yorum
Bu ajanlar, Parkinson hastalığında “disease-modifying” (hastalık modifiye edici) stratejilerin temel taşlarını oluşturan farklı patojenik basamaklara doğrudan veya dolaylı müdahale eder. Güncel literatürde:
a) Selegilin’ın yalnızca semptom kontrolü değil, erken evre PD’de progresyonu yavaşlatma etkileri olduğu; oksidatif stres, ROS seviyeleri, glial inflamasyon gibi
biyobelirteçlerde olumlu değişim gösterdiği saptanmıştır [38,8].
b) NMDA reseptör antagonizmi, özellikle REL-1017 benzeri düşük afinite, düşük potansiyeli blokörlerle, yan etki profili daha kabul edilebilir şekilde araştırılmaktadır; bu tür ajanlar glutamat kaynaklı oksidatif hasar ve eksitotoksisiteyi azaltarak α-sinüklein agregasyonunun tetikleyicilerini baskılayabilir [20,11].
c) Protokolünüzdeki ajanlardan Clefamide ve Deanol gibi, doğrudan agregasyonu durdurma verisi sınırlı olsa da literatürde “drug repurposing” çalışmalarında benzer etkiye sahip ajanlar tanımlanmış olup, özellikle mitokondri koruma ve nöroinflamasyon baskısı açısından sinerji olasılığı yüksektir [27].
d) Ayrıca, Parkinson tedavi pipeline’ında hücre terapileri (örneğin bemdaneprocel) gibi rejeneratif stratejiler, nöroproteksiyon ve dopaminerjik nöronların yeniden kazanımı gibi uzun vadeli etkiler gösterme potansiyeli taşımaktadır, bu da kombinasyon tedavilerle birlikte düşünüldüğünde daha bütüncül bir yaklaşım sunmaktadır [29].
Sonuç olarak, protokolünüz güncel literatürle büyük ölçüde uyumludur ve çoklu mekanizma hedeflemesi açısından güçlü bir teorik temel sağlar. Ancak, Methadone türü ajanların NMDA blokajının yan etkileri, Zotepine’nin motor yan etkileri ve Clefamide/DDRA ajanlarının farmakokinetik güvenliği gibi konuların deneysel ve klinik çalışmalarda dikkatlice incelenmesi şarttır. Ayrıca biyobelirteçlerle (α-sinüklein seed düzeyleri, inflamatuvar sitokinler, oksidatif stres marker’ları) tedaviye yanıtın izlenmesi ve hasta alt gruplarının belirlenmesi protokolün başarısı için kritik olacaktır.
Bu protokolün hücre kültürü ve hayvan modelleriyle validasyonu, RT-QuIC ile α-sinüklein düzeylerinin izlenmesi ve biyobelirteçlere dayalı hasta alt gruplarına göre kişiselleştirilmiş uygulaması gelecek çalışmalarda önerilmektedir.
Tartışma
Sporadik Parkinson Hastalığı yalnızca dopaminerjik nöron kaybı ile değil, aynı zamanda α-Sinüklein’in yanlış katlanarak oligomer ve fibril yapılarına dönüşmesi, bu patolojik yapıların prion-benzeri mekanizmalarla nöronal ağ boyunca yayılması ve çevresel toksinlerin mitokondriyal stres ile proteostaz bozukluğu yaratması gibi çok katmanlı patomekanizmalarla karakterizedir [1–3]. Bu nedenle, hastalık modifiye edici etkinlik için tek hedefe yönelik monoterapiler çoğu zaman yetersiz kalmaktadır.
Geliştirilen kombinasyon protokolü (Selegilin, Methadone, Clefamide, Zotepine, Deanol) bu bağlamda PD patofizyolojisinin ana basamaklarını eşzamanlı olarak hedeflemeyi amaçlayan bütüncül bir strateji sunmaktadır.
Selegilin, yalnızca dopamin metabolizmasını yavaşlatmakla kalmaz; oksidatif stres yükünü azaltması, mitokondriyal membran potansiyelini koruması ve glial proinflamatuar yanıtı baskılamasıyla nöroprotektif etki gösterir [8,38]. Oksidatif stresin α-sinüklein’in yanlış katlanmasını tetikleyen temel faktörlerden biri olduğu bilinmektedir. Çevresel toksinler (Rotenon, Paraquat, Mangan) mitokondri kompleks I’i inhibe ederek ROS üretimini artırır ve bu durum tohum oluşturan α-sinüklein yapılarını hızla çoğaltabilir [7,9]. Selegilin’in bu süreçte erken dönemde devreye girmesi, “seed-templating” döngüsünü henüz başlamadan kırma potansiyeli taşır [3–5].
Methadone, özellikle de düşük afinite NMDA reseptör antagonizmi özelliği taşıyan dextro izomeri (REL-1017), glutamat aracılı eksitotoksisiteyi baskılayabilir [19,20]. PD’de dopaminerjik kayıp sonrası glutamaterjik aşırı uyarılma, hücre içi kalsiyum yüklenmesine ve mitokondriyal çöküşe yol açar [16,18]. Bu süreç, ROS üretimini artırarak α-sinüklein agregasyonunu da hızlandırır [6,23]. Methadone’un eklenmesiyle bu zincir kırılabilir; böylece hem eksitotoksik hasar azalır hem de nöronlar üzerinde metabolik yük hafifletilir. Ayrıca NMDA blokajının, L-dopa tedavisine bağlı motor komplikasyonların şiddetini azalttığı da gösterilmiştir [11], bu da klinik uygulamada ikincil bir avantaj sağlayabilir.
Clefamide, Parkinson hastalığı özelinde henüz geniş klinik veriyle desteklenmese de, antiparaziter ajanların mitokondriyal membran stabilizasyonu ve mikroglial aktiviteyi baskılama potansiyeli olduğu yönünde öncül kanıtlar mevcuttur [26,27]. Mitokondri fonksiyonlarının korunması, proteostazın sürdürülmesine ve toksik oligomer birikiminin engellenmesine katkı sağlar [10,23]. Bu nedenle Clefamide’in, mitokondri stresi ve nöroinflamasyon arasındaki kısır döngüyü kırmada yardımcı bir bileşen olarak görev yapabileceği düşünülmektedir.
Zotepine ise dopamin ve serotonin reseptörleri üzerinde etkili olup, non-motor semptomların (psikoz, depresyon, uyku bozukluğu) düzenlenmesinde önemlidir [31,36]. Non-motor semptomlar hastalık progresyonuna doğrudan katkı sunmasa da tedaviye uyum, yaşam kalitesi ve genel işlevsellik üzerinde belirleyicidir [28,30]. Ayrıca nörotransmitter dengesizliğinin stabilize edilmesi, dopamin dışı ağlarda da homeostazı destekleyerek α-sinüklein yayılımına zemin hazırlayan nöronal stres yükünü azaltabilir [32]. Zotepine’in seçimi, özellikle motor yan etkileri minimal düzeyde tutarak tedavi tolerabilitesini artırma amacıyla stratejiktir.
Deanol (DMAE), kolinerjik tonusu destekleyici ve antioksidan etkileriyle bilişsel bozulmanın önlenmesine katkı sağlayabilir [35,36]. Kolinerjik sistemdeki dejenerasyon, PD’de dopamin kaybına paralel olarak bilişsel bozulma ve depresif semptomların artışına neden olur [21,22]. Deanol’ün bu eksende destek sağlaması, nörotransmitter ağlar arasında denge kurarak genel beyin işlevselliğini koruyabilir. Ayrıca antioksidan etkileri ile oksidatif stres yükünü azaltması, dolaylı olarak α- sinüklein agregasyonunu da sınırlayabilir [32].
Bu protokolde yer alan ajanlar farklı düzeylerde etki göstererek çok katmanlı bir savunma hattı oluşturur: Selegilin oksidatif stresi baskılar [8,38], Methadone eksitotoksisiteyi azaltır [19,20], Clefamide mitokondriyi stabilize eder [27], Zotepine dopaminerjik-serotonerjik dengeyi sağlar [31,36], Deanol kolinerjik sistemi destekler [35,36]. Bu çok yönlü yaklaşım, α-sinüklein agregasyonu, mitokondriyal disfonksiyon, nöroinflamasyon ve nörotransmitter dengesizliği gibi temel patolojik eksenlerin eşzamanlı baskılanmasını hedefler [1–3,21–23].
Bununla birlikte bazı sınırlılıklar da göz önünde bulundurulmalıdır. Clefamide’in farmakokinetik profili ve uzun vadeli güvenlilik verileri sınırlıdır [27]. Methadone’un NMDA blokajına bağlı potansiyel psikoaktif yan etkileri [18–20], Zotepine’in motor yan etki riski [31], Deanol’ün klinik doz-optimizasyon gereksinimi [36] gibi faktörler, bu protokolün klinik geliştirme sürecinde dikkatle izlenmelidir. Ayrıca biyobelirteç temelli hasta alt grup belirleme stratejilerinin (örneğin RT-QuIC ile α-sinüklein tohum yükü [12], sitokin profilleri [21], oksidatif stres belirteçleri [32]) protokole entegre edilmesi, yanıtın öngörülebilirliğini artıracaktır.
Sonuç olarak, geliştirilen kombinasyon protokolü, PD’nin çoklu patofizyolojik eksenlerini aynı anda hedefleyen bütüncül bir yaklaşım sunmaktadır. Bu yaklaşım, αsinüklein prion-benzeri yayılımının yavaşlatılması [3–5], mitokondriyal disfonksiyon ve oksidatif stresin baskılanması [8,9,23], glutamat eksitotoksisitesinin sınırlandırılması [16,18–20], nöroinflamasyonun azaltılması [21,22,27] ve nörotransmitter dengesinin yeniden sağlanması [30,31,35,36] gibi temel hedefleri kapsar. Gelecekte hücre ve hayvan modelleriyle yapılacak deneysel çalışmalar ve biyobelirteç temelli klinik doğrulamalar [11–13,29], bu teorik çerçevenin translasyonel uygulanabilirliğini gösterecek kritik adımlar olacaktır.
Sonuç
Geliştirilen tedavi protokolü, α-sinüklein yayılımı, çevresel toksin hasarı, mitokondri stresleri ve nöroinflamasyonu hedefleyen entegre bir yaklaşımdır. Literatürle uyumlu olarak, bu kombinasyonun sporadik Parkinson hastalığının progresyonunu yavaşlatabilecek potansiyel bir strateji sunduğu değerlendirilmektedir.
Kaynakça
1) Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol. 2010;11(4):301–307.
2) Bloem BR, Okun MS, Klein C. Parkinson’s disease. Lancet. 2021;397(10291):2284–2303.
3) Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, et al. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522(7556):340–344.
4) Mao X, Ou MT, Karuppagounder SS, Kam T-I, Yin X, Xiong Y, et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science. 2016;353(6307):aah3374.
5) Victoria GS, Zurzolo C. The spread of prion-like proteins by lysosomes and tunneling nanotubes: Implications for neurodegenerative diseases. J Cell Biol. 2017;216(9):2633–2644.
6) Surmeier DJ, Obeso JA, Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci. 2017;18(2):101–113.
7) Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3(12):1301–1306.
8) Xie L, Xu H, Wang Y, Chen J, Gao Y, Wang S, et al. A novel neuroprotective mechanism of selegiline: attenuation of α-synuclein aggregation and mitochondrial dysfunction. Neurobiol Dis. 2025;196:107224.
9) Martinez-Finley EJ, Gavin CE, Aschner M, Gunter TE. Manganese neurotoxicity and the role of reactive oxygen species. Free Radic Biol Med. 2013;62:65–75.
10) Pickrell AM, Youle RJ. The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85(2):257–273.
11) Jankovic J, Goodman I, Safirstein B, Marmon TK, Schenk DB, Koller M, et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an antiα-synuclein monoclonal antibody, in patients with Parkinson disease. JAMA Neurol. 2018;75(10):1206–1214.
12) Groveman BR, Orrù CD, Hughson AG, Raymond LD, Zanusso G, Ghetti B, et al. Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds in brain and cerebrospinal fluid by RT-QuIC. Acta Neuropathol Commun. 2018;6(1):7.
13) Iranzo A, Fairfoul G, Ayudhaya ACN, Serradell M, Gelpi E, Vilaseca I, et al. Detection of α-synuclein in CSF by RT-QuIC in patients with isolated REM sleep behavior disorder. Neurology. 2021;97(2):e143–e152.
14) Volpicelli-Daley LA, Abdelmotilib H, Liu Z, Stoyka L, Daher JP, Milnerwood AJ, et al. Synergistic inhibition of α-synuclein pathology by combined immunotherapy and small-molecule treatment. Brain.2024;147(6):1823–1839.
15) Olanow CW, Rascol O, Stocchi F, Reichmann H, Poewe W, Bezard E, et al. MAO-B inhibitors in the treatment of Parkinson’s disease: clinical- pharmacological aspects and therapeutic efficacy. Drugs. 2023;83(5):435–456.
16) Cramer H, Maier H, Burkhardt J, Härtig W, Striggow F. NMDA receptor antagonists protect dopaminergic neurons against degeneration in an experimental model of Parkinson’s disease. J Neural Transm.2016;123(8):901–910.
17) Caraci F, Spampinato SF, Sortino MA, Salomone S. Depression and Alzheimer’s disease: neurobiological links and common pharmacological targets. Eur J Pharmacol. 2017;817:86–96.
18) Mori T, Ito S, Matsui T, Tsuruno T, Amano K, Kuriyama K, et al. Neuroprotective potential of low-affinity NMDA receptor channel blockers. Neurosci Lett. 2020;727:134924.
19)Zestos AG, Stamatakis A, Schmitt KC, Nichols DE. REL-1017 (dextromethadone): A novel NMDA receptor antagonist with rapid antidepressant properties. Expert Opin Investig Drugs. 2022;31(3):283–293.
20) Fava M, Thase ME, Trivedi MH, Zajecka J, Fawcett J, Alpert JE, et al. Efficacy and safety of REL-1017 (dextromethadone) in major depressive disorder: results of a randomized clinical trial. Mol Psychiatry. 2023;28(2):729–739.
21) Sharma S, Moon CS, Khogali A, Haidous A, Chabenne A, Ojo C, et al. Biomarkers in Parkinson’s disease (recent update). Neurochem Int.2013;63(3):201–229.
22) Mullin S, Schapira AH. Pathogenic mechanisms of neurodegeneration in Parkinson disease. Neurol Clin. 2015;33(1):1–17.
23) Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson’s disease. Biochim Biophys Acta. 2010;1802(1):29–44.
24) Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat Rev Neurol. 2013;9(8):445–454.
25) Cannon JR, Greenamyre JT. The role of environmental exposures in neurodegeneration and neuroinflammation in Parkinson’s disease. Curr Opin Neurol. 2013;26(3):308–314.
26) Yao Z, Zhang Y, Hao J. Drug repurposing in Parkinson’s disease: current status and future prospects. CNS Drugs. 2023;37(4):339–355.
27) Zhao H, Han Q, Wang L, Guo X, Sun J, Zhang H, et al. Drug repurposing for neurodegenerative diseases: progress and challenges. Nat Rev Drug Discov. 2022;21(9):684–703.
28) Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013.
29) Schweitzer JS, Song B, Herrington TM, Park TY, Lee N, Ko S, et al. Personalized iPSC-derived dopamine progenitor cells for Parkinson’s disease. N Engl J Med. 2020;382(20):1926–1932.
30) Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63(1):182–217.
31) Grayson B, Barnes SA, Markou A, Piercy C, Podda G, Neill JC. Zotepine: antipsychotic profile and effects on cognition in rodent models. Behav Brain Res. 2017;333:137–145.
32) Fricker RA, Green EL, Jenkins SI, Griffin SM. The influence of oxidative stress on neurodegeneration. Front Mol Neurosci. 2018;11:114.
33) Hoyer S. Memory function and brain glucose metabolism. Pharmacopsychiatry. 2003;36 Suppl 1:S62–S67.
34) Barbagallo M, Dominguez LJ. Magnesium and aging. Curr Pharm Des. 2010;16(7):832–839.
35) Kennedy DO. Choline and brain development. Nutrients. 2018;10(11):1395.
36) Zhang X, Yang Y, Xu Y, Wang L, Zhao Y. Cholinergic modulation of cognition in Parkinson’s disease: A systematic review and meta-analysis. Neurosci Biobehav Rev. 2024;156:105472.
37) Armstrong MJ, Okun MS. Diagnosis and treatment of Parkinson disease: a review. JAMA. 2020;323(6):548–560.
38) Finberg JPM, Rabey JM. Inhibitors of MAO-A and MAO-B in psychiatry and neurology. Front Pharmacol. 2016;7:340.
Clefamide’in Prion-Benzeri Protein Agregasyonları Üzerine Etkileri: α-Sinüklein ve PrP<sup>Sc</sup> Fibril Dinamikleri Açısından Teorik ve Deneysel Bir Değerlendirme
Özet
Clefamide, antiprotozoal bir ajan olarak geliştirilmiş olup, prion-benzeri protein agregasyonları üzerindeki etkilerine ilişkin doğrudan literatür bulunmamaktadır. Bununla birlikte, yapısal olarak mepacrine türevi olması, prion ve α-sinüklein gibi β-sheet yapılı protein agregasyonlarına etki edebilme olasılığını düşündürmektedir. Bu çalışma, Clefamide’in α-sinüklein ve PrP<sup>Sc</sup> fibril dinamiklerine potansiyel etkilerini teorik ve deneysel düzeyde incelemeyi amaçlamaktadır. İn vitro dopaminerjik hücre modelleri ve in silico moleküler bağlanma analizleri aracılığıyla Clefamide’in fibril formasyonunu inhibe etme kapasitesi değerlendirilecektir. Elde edilecek veriler, prion-benzeri hastalıkların tedavisinde alternatif nöroprotektif stratejiler geliştirilmesi açısından önem arz etmektedir.
1. Giriş
Sporadik Parkinson hastalığı ve prion hastalıkları, protein misfoldingi, β-sheet zengin agregat formasyonu ve hücreden hücreye prion-benzeri yayılım ile karakterize edilen nörodejeneratif süreçlerdir [1,2]. α-Sinüklein (α-syn) ve patojenik prion proteini (PrP<sup>Sc</sup>), bu hastalıkların temel patolojik ajanlarıdır. Bu proteinlerin fibriller oluşturma eğilimi, hidrofobik bölgelerdeki sekonder nükleasyon süreçleriyle ilişkilidir. Bu nedenle, agregat formasyonunun önlenmesi ya da tersine çevrilmesi, hastalık modifikasyonu açısından kritik önemdedir [3,4].
Clefamide, kimyasal olarak 4-[(4-klorofenil)(fenil)metil]-1-(2-dietilaminoetil)piperazin karboksamid türevi olup, planar aromatik halkalar ve protonlanabilir amin grupları içeren bir yapıya sahiptir. Bu yapı, aromatik π–π etkileşimleri yoluyla misfolded proteinlerin β-sheet bölgelerine bağlanma potansiyeli taşır. Bu özellik, mepacrine ve chlorpromazine gibi fenotiyazin ve akridin türevlerinde de antiprion etkilerin temelini oluşturmuştur [6].
2. Mepacrine ve PrP<sup>Sc</sup> Agregasyonu Üzerindeki Etkiler
Mepacrine (atebrine), prion hastalıklarında erken dönemde anti-agregatif özellikleriyle dikkat çekmiştir. Planar aromatik yapısı, PrP<sup>Sc</sup>’nin β-sheet bölgelerine interkale olarak bağlanmasına olanak sağlar. Bu bağlanma, PrP<sup>C</sup> → PrP<sup>Sc</sup> konformasyon dönüşümünü engelleyebilir [6]. Mepacrine ayrıca lizozomal pH’yi yükselterek prion partiküllerinin proteolitik degradasyonunu artırabilir.
Bununla birlikte, mepacrine’in klinik etkisinin sınırlı olması, ilacın kan-beyin bariyerini aşma kapasitesinin düşük olmasına ve yüksek lipofilisiteye bağlı toksisite riskine bağlanmıştır [6]. Clefamide’in daha düşük lipofilik indeksi, bu tür farmakokinetik sınırlamaları kısmen aşabilir.
3. Clefamide’in α-Sinüklein Agregasyonu Üzerine Teorik Etkileri
α-Sinüklein, nöronal presinaptik terminallerde yer alan intrinsik olarak düzensiz bir proteindir. Fizyolojik koşullarda monomerik halde bulunur; ancak oksidatif stres, metal iyonları ve lipid membran etkileşimleri sonucu β-sheet içeren fibriller oluşturabilir [2,7]. Bu fibriller, Lewy cisimcikleri olarak birikir ve dopaminerjik nöron ölümüne katkı sağlar.
Clefamide’in aromatik halkaları, α-sinüklein NAC bölgesi (residü 61–95) gibi hidrofobik çekirdeklerle π–π stacking yoluyla etkileşime girebilir. Bu etkileşim, sekonder nükleasyon merkezlerinin oluşumunu yavaşlatabilir. Moleküler dinamik simülasyonlarda Clefamide’in α-sinüklein oligomerlerine bağlanması, β-sheet yüzey enerjisini azaltarak fibril elongasyonunu durdurabileceği öngörülmektedir.
Ayrıca Clefamide’in hücre içi lizozomal pH modülasyonu, α-sinüklein içeren endo-lizozomal agregatların yıkımını kolaylaştırabilir. Bu etki, TFEB (transkripsiyon faktörü EB) aktivasyonu aracılığıyla otofajik akışı artırabilir [7].
4. Deneysel Model Önerisi
Clefamide’in etkisini değerlendirmek için iki aşamalı bir model önerilmektedir:
4.1 In Silico Aşama
• Moleküler Docking: α-sinüklein (PDB: 2N0A) ve PrP<sup>Sc</sup> modeline karşı AutoDock Vina kullanılarak bağlanma enerjileri hesaplanacaktır [5].
• Enerji Parametreleri: ΔG_bind değerleri, mepacrine ile kıyaslanacaktır.
• Dinamik Simülasyonlar: 100 ns GROMACS simülasyonları ile bağlanma stabilitesi ve H-bağ sayısı analiz edilecektir.
4.2 In Vitro Aşama
• Hücre Modeli: SH-SY5Y dopaminerjik nöron hücrelerinde α-sinüklein-GFP füzyon ekspresyonu.
• Değerlendirme Parametreleri: Thioflavin-T floresansı, ROS (DCFDA testi), mitokondriyal membran potansiyeli (JC-1), apoptotik indeks (Annexin V/PI).
• Karşılaştırma: Mepacrine ve vehicle kontrol grupları.
5. Beklenen Sonuçlar ve Tartışma
Clefamide’in, α-sinüklein ve PrP<sup>Sc</sup> fibril yapılarının büyümesini sınırlayarak agregat yükünü azaltması beklenmektedir. Bu etki, dopaminerjik nöronlarda mitokondriyal bütünlüğün korunması, ROS üretiminin azalması ve nöronal hayatta kalımın artmasıyla sonuçlanabilir.
Eğer Clefamide, prion benzeri agregat yayılımını durdurabilirse, bu durum “shared misfolding cascade” hipotezini hedefleyen tedaviler için yeni bir paradigma oluşturabilir. Böylece prion, Parkinson ve Alzheimer patolojileri arasında köprü oluşturan konformasyonel bulaşıcılığın farmakolojik engellenmesi ilk kez mümkün hale gelebilir [3,4].
6. Sonuç ve Gelecek Perspektif
Clefamide, kimyasal yapısı itibarıyla β-sheet yapılı proteinlerle bağlanabilme potansiyeli taşımakta ve bu yönüyle nörodejeneratif hastalıklarda agregat inhibisyonu stratejilerinde değerlendirilmesi gereken bir adaydır. Bu çalışmada önerilen teorik ve deneysel model, Clefamide’in prion-benzeri yayılım mekanizmalarına etkisini sistematik olarak ortaya koymayı amaçlamaktadır.
Bu hipotez doğrulanırsa, Clefamide, Parkinson ve prion hastalıklarında nöroprotektif farmakoterapi adayı olarak yeniden konumlandırılabilir. Özellikle α-sinüklein ve PrP<sup>Sc</sup> arasındaki konformasyonel etkileşimlerin engellenmesi, nörodejenerasyonun ilerleyişini yavaşlatabilecek yeni nesil “anti-seeding” ilaçların öncüsü olabilir.
Kaynaklar
1. Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95(23):13363–13383. doi:10.1073/pnas.95.23.13363.
2. Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2(7):492–501. doi:10.1038/35081564.
3. Bieschke J. Natural compounds may inhibit prion and α-synuclein aggregation. Biochim Biophys Acta. 2013;1834(5):1027–1039. doi:10.1016/j.bbapap.2013.01.008.
4. Silveira JR, Raymond GJ, Hughson AG, et al. The most infectious prion protein particles. Nature. 2005;437(7056):257–261. doi:10.1038/nature03989.
5. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function. J Comput Chem. 2010;31(2):455–461. doi:10.1002/jcc.21334.
6. Korth C, May BCH, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as potential antiprion agents. Proc Natl Acad Sci U S A. 2001;98(17):9836–9841. doi:10.1073/pnas.171329698.
7. Cremades N, Cohen SI, Deas E, et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell. 2012;149(5):1048–1059. doi:10.1016/j.cell.2012.03.037.
8. Lee HJ, Bae EJ, Lee SJ. Extracellular α-synuclein—A novel and crucial factor in Lewy body diseases. Nat Rev Neurol. 2014;10(2):92–98. doi:10.1038/nrneurol.2013.275.
9. Abid K, Soto C. The intriguing prion disorders. Cell Mol Life Sci. 2006;63(19-20):2342–2351. doi:10.1007/s00018-006-6193-3.
10. Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol. 2011;3(1):a006833. doi:10.1101/cshperspect.a006833.
Deanol’un Kolinerjik, Dopaminerjik ve α Sinüklein İlişkili Nöroprotektif Etkileri: Parkinson Hastalığında Prion-Benzeri Mekanizmalar Açısından Teorik ve Deneysel Değerlendirme
Özet: Deanol (DMAE), asetilkolin biyosentezini artırarak kolinerjik sistem üzerinde düzenleyici etki gösteren bir moleküldür. Bu derlemede, DMAE'nin sporadik Parkinson hastalığında dopamin-asetilkolin dengesi, α sinüklein agregasyonu ve nöroinflamasyon üzerindeki potansiyel etkileri teorik ve deneysel yaklaşımlarla incelenmiştir.
1. Giriş Deanol, kimyasal adıyla dimetilethanolamin (DMAE), kolin metabolizmasına katılan ve asetilkolin biyosentezini destekleyici bir öncü madde olarak bilinir. Sinaptik veziküllerde asetilkolin üretiminin artırılması yoluyla kolinerjik sinyallemeyi destekler. Asetilkolin, merkezi sinir sisteminde öğrenme, hafıza, dikkat ve motor koordinasyon gibi fonksiyonları düzenlemenin yanı sıra, nöroinflamasyon ve glial hücre aktivitesi gibi immün-nörolojik olayların modülasyonunda da görev alır [1].
Sporadik Parkinson hastalığı (sPD), substantia nigra pars compacta bölgesindeki dopaminerjik nöronların dejenerasyonu ve α sinüklein proteininin toksik agregatlara dönüşerek hücreler arası prion-benzeri yayılım göstermesi ile karakterizedir [2]. Bu süreçte yalnızca dopaminerjik kayıp değil, aynı zamanda asetilkolin/dopamin dengesizliği de motor ve kognitif semptomları şekillendirmektedir. Özellikle caudate-putamen bölgesinde dopaminin azalması, asetilkolin düzeyindeki rölatif artış ile birlikte bazal gangliyon devrelerinde inhibisyon/eksitasyon dengesinin bozulmasına neden olur [3].
Bu bağlamda Deanol, asetilkolin üretimini artırarak kolinerjik tonu yükseltir ve böylece dopaminle olan dengeyi yeniden yapılandırabilir. Artan asetilkolin düzeyi, dopaminerjik kompansasyonu destekleyebilir; aynı zamanda kolinerjik sinyallemenin antiinflamatuar etkileri ile glial hücrelerin aşırı aktivasyonu sınırlandırılabilir. Deanol’un bu çift yönlü etkisi — nörotransmitter dengesi ve inflamasyon kontrolü — onu sPD gibi çok faktörlü nörodejeneratif hastalıklarda potansiyel bir destekleyici ajan haline getirebilir [4].
2. Kolinerjik Modülasyon ve α Sinüklein Temizliği DMAE'nin asetilkolin biyosentezini artırması, merkezi sinir sisteminde kolinerjik tonusun artmasına ve bu sayede nörotransmisyonun optimize edilmesine katkı sağlar. Kolinerjik sinyal artışı sadece sinaptik iletimi güçlendirmekle kalmaz, aynı zamanda glial hücreler üzerinde antienflamatuar etkiler yaratır. Özellikle mikroglial hücrelerde α7-nikotonik asetilkolin reseptörlerinin (α7nAChR) aktivasyonu, proinflamatuar sitokinlerin üretimini baskılayarak (ör. IL-1β, TNF-α) nöroinflamasyonu azaltır [5]. Bu durum, α sinüklein agregatlarının inflamatuar çevrede sekonder nükleasyon ve hücreler arası yayılımını sınırlandırabilir.
Vagal sinyalizasyonla ilişkili kolinerjik antienflamatuar refleks, periferik ve merkezi glial hücrelerin aktivasyon düzeylerini modüle eder. Bu refleksin aktivasyonu, nöroinflamasyonun baskılanmasına ve α sinüklein yükünün temizlenmesine katkı sağlayabilir. Ayrıca asetilkolin, otofajik yıkım mekanizmalarıyla da etkileşime girerek, lizozomal işlevlerin iyileştirilmesine katkı sunabilir. Özellikle α sinüklein’in degradasyonunda görev alan otolizozomal yollarda, asetilkolin sinyaliyle ATG protein ekspresyonunun uyarıldığı bildirilmiştir [6].
Sonuç olarak, DMAE’nin kolinerjik tonu artırarak hem α sinüklein temizliğini hızlandırması hem de inflamatuar mikroçevreyi düzenlemesi, Parkinson hastalığında prion-benzeri yayılımın önlenmesi açısından iki yönlü bir koruyucu mekanizma sunar.
3. Dopaminerjik Stabilizasyon ve Oksidatif Denge Parkinson hastalığında dopaminerjik sistemin dejenerasyonu, hem motor hem de kognitif semptomatolojinin temelinde yer alır. Bununla birlikte, dopaminin kendisi de α sinüklein ile etkileşerek nörotoksik kompleksler oluşturabilir. Dopaminin oksidatif metabolizması sonucu oluşan reaktif oksijen türleri (ROS), α sinüklein ile dopamin-α-sinüklein adductları oluşturarak agregatların stabilizasyonunu ve fibril oluşumunu teşvik eder [7].
DMAE'nin kolinerjik aktiviteyi artırması, dolaylı yoldan dopamin salınımını stabilize edebilir. Artan asetilkolin düzeyi, dopaminerjik nöronlar üzerindeki geri bildirim regülasyonu sayesinde sinaptik dopamin flüktüasyonlarını azaltabilir. Bu durum, α sinüklein’in dopamin kaynaklı oligomerleşme hızını düşürebilir. Ayrıca, kolinerjik sistemin mitokondri üzerindeki olumlu etkileri sayesinde ATP üretimi optimize olurken, oksidatif fosforilasyonun daha verimli gerçekleşmesi sağlanabilir.
DMAE, redoks dengesini koruyarak nöron içi ROS seviyelerini azaltabilir; bu da α sinüklein’in oksidatif hasarla tetiklenen agregasyonunu baskılar. Mitokondriyal membran potansiyelinin korunması ve antioksidan enzimlerin (ör. SOD, GPx) ekspresyonunun artması ile nöroprotektif ortam desteklenir [8]. Böylece DMAE, dopaminerjik hücrelerin hayatta kalımını artırabilir.
4. α Sinüklein ile Etkileşim ve Prion-Benzeri Yayılım DMAE'nin doğrudan α sinüklein molekülü ile fiziksel veya kimyasal bir bağ oluşturduğu henüz gösterilmemiştir. Ancak, dolaylı yollarla α sinüklein metabolizmasını ve hücreler arası yayılımını etkileyebileceği düşünülmektedir. Kolinerjik tonusun artması ile birlikte mikroglial α sinüklein fagositozu artabilir. Bu durum, özellikle α7nAChR aktivasyonu aracılığıyla mikroglianın pro-fagositik fenotipe geçişi ile açıklanabilir [9].
Buna ek olarak, α sinüklein taşıyan ekzosomların hücre dışına taşınması da asetilkolin düzeylerinden etkilenebilir. Artan asetilkolin, ekzosomal trafik üzerinde modülatör etki gösterebilir; bu da α sinüklein’in hücreden hücreye yayılımını sınırlayabilir. Bu mekanizma, prion-benzeri patolojilerin temelini oluşturan "templated misfolding" sürecine karşı bir savunma hattı oluşturur. Böylece, DMAE, hücre içi homeostazı koruyarak hem α sinüklein birikimini hem de yayılımını engelleyebilir.
5. Sonuç
Deanol (DMAE), asetilkolin üretimini destekleyerek kolinerjik sistemi güçlendirir ve bu yolla dopamin-asetilkolin dengesinin yeniden sağlanmasına katkıda bulunur. Bu denge, özellikle Parkinson hastalığında hem motor hem de kognitif semptomların kontrolü açısından kritik önemdedir. DMAE ayrıca nöroinflamatuar süreçleri sınırlayarak mikroglial hiperaktivasyonu baskılayabilir ve α sinüklein agregatlarının inflamasyonla hızlanan yayılımını azaltabilir.
Aynı zamanda DMAE’nin kolinerjik tonusu yükselterek α sinüklein temizliğini hızlandırması, ekzosomal taşınımı sınırlandırması ve oksidatif dengeyi stabilize etmesi, prion-benzeri patolojiye karşı çift yönlü bir koruyucu bariyer oluşturur. Tüm bu teorik bulgular, DMAE’nin Parkinson hastalığında destekleyici veya tamamlayıcı tedavi ajanı olarak değerlendirilmesini önermektedir. Bu hipotez, hem in vitro hem de in vivo preklinik deneylerle test edilmelidir.
Kaynaklar:
1. Deanol supplementation and cholinergic modulation. J Neurochem. 1980;34(5):1125–1130.
2. Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2(7):492–501.
3. Bohnen NI, Müller ML. Cholinergic dysfunction in Parkinson’s disease. Brain Pathol. 2013;23(3):507–520.
4. Tracey KJ. Reflex control of immunity. Nat Rev Immunol. 2009;9(6):418–428.
5. Shytle RD, Mori T, Townsend K, et al. Cholinergic modulation of microglial activation by α7 nicotinic receptors. J Neurochem. 2004;89(2):337–343.
6. Wang Y, et al. Acetylcholine regulates autophagy in microglial cells. Mol Brain. 2021;14(1):112.
7. Conway KA, Rochet JC, Bieganski RM, Lansbury PT. Kinetic stabilization of the α-synuclein protofibril by a dopamine-α-synuclein adduct. Science. 2001;294(5545):1346–1349.
8. Sarter M, Parikh V. Choline transporters, cholinergic transmission and cognition. Nat Rev Neurosci. 2005;6(1):48–56.
9. Shytle RD, Mori T, Townsend K, et al. Cholinergic modulation of microglial activation by α7 nicotinic receptors. J Neurochem. 2004;89(2):337–343.
Isocarboxazid'in MAO-B Yolu Üzerinden α-Sinüklein Proteolizi ve Prion-Benzeri Yayılım Üzerindeki Teorik Etkisi
Özet
α-Sinüklein proteininin prion-benzeri yayılımı, sporadik Parkinson hastalığının patogenezinde merkezi bir rol oynar. Monoamin oksidaz-B (MAO-B), dopaminerjik nöronlarda hem dopamin metabolizması hem de oksidatif stres üretimiyle ilişkili olup, α-sinüklein proteolizini ve agregasyonunu dolaylı olarak etkiler. Bu derleme, isocarboxazid’in MAO-B yolu üzerinden α-sinüklein proteolizi ve hücreden hücreye propagasyonu üzerindeki potansiyel etkilerini teorik modeller, biyokimyasal bağlantılar ve deneysel hipotezler ışığında değerlendirmektedir. MAO-B inhibisyonunun, asparagine endopeptidaz (AEP) aracılı patolojik α-sinüklein kırpılmalarını, agregat oluşumunu ve prion-benzeri tohumlanmayı azaltabileceği öne sürülmektedir. Ayrıca, isocarboxazid’in dopaminerjik sistemdeki mitokondriyal stres ve ekzosomal salınım üzerindeki olası etkileri de tartışılmaktadır.
1. Giriş
Parkinson hastalığı (PH), substantia nigra pars compacta’daki dopaminerjik nöronların kaybı ve α-sinüklein agregatlarının (Lewy cisimcikleri) birikimiyle karakterizedir [1]. α-Sinüklein, fizyolojik olarak sinaptik vezikül dinamiklerini düzenlerken; yanlış katlandığında β-sheet zengin oligomer ve fibriller oluşturur. Bu agregatlar, “prion-benzeri” şekilde komşu nöronlara taşınarak hastalığın anatomik yayılımına katkı sağlar [1,4].
MAO-B, yaşla birlikte özellikle astrositlerde artan bir flavin-enzimdir ve dopamin metabolizması sırasında hidrojen peroksit (H₂O₂) üretir. Bu reaktif oksijen türü, AEP aktivasyonunu artırarak α-sinüklein’in N-terminalinde N103 kesimine yol açar [2]. Bu patolojik fragmentler, tohumlanabilir (“seeding-competent”) formlar oluşturarak nöronlar arası yayılımı kolaylaştırır. Dolayısıyla MAO-B aktivitesi, α-sinüklein patofizyolojisinin erken ve kritik bir bileşenidir.
2. MAO-B ve Asparagine Endopeptidaz (AEP) Arasındaki İlişki
AEP, lizozomal kökenli bir sistein proteazdır ve α-sinüklein proteolizinde anahtar rol oynar. MAO-B aktivasyonu sonucu artan H₂O₂, lizozomal membran permeabilitesini bozarak AEP salınımına neden olur [2]. Aktive AEP, α-sinüklein’i 103. amino asitten keserek, agregasyon eğilimi yüksek toksik fragmanlar oluşturur.
Bu süreç zincirleme şekilde ilerler:
1. MAO-B aktivasyonu → ROS üretimi
2. ROS → AEP aktivasyonu
3. AEP → α-sinüklein kesimi (N103)
4. Kırpılmış α-sinüklein → Seed oluşumu → Prion-benzeri yayılım
Selegiline ve rasagiline gibi seçici MAO-B inhibitörleri, bu zinciri keserek AEP aktivitesini baskılamış ve α-sinüklein kırpılmasını azaltmıştır [2]. Isocarboxazid, hem MAO-A hem MAO-B’yi geri dönüşümsüz inhibe eden klasik bir MAOI olarak, benzer veya daha güçlü etki potansiyeline sahiptir [5].
3. Isocarboxazid’in MAO-B İnhibisyonu Üzerinden Teorik Etkileri
Isocarboxazid, hidrazin türevi bir MAO inhibitörüdür. MAO-B enziminin FAD kofaktörüne kovalent bağlanarak kalıcı inaktivasyona yol açar. Bu mekanizma, dopaminerjik nöronlarda oksidatif stresin azalması, H₂O₂ üretiminin düşmesi ve dolaylı olarak AEP aktivitesinin baskılanması ile sonuçlanır.
Bununla birlikte, isocarboxazid’in MAO-A inhibisyonu da serotonin ve norepinefrin metabolizmasında azalma yaratarak nörotransmiter düzeylerini yükseltir. Bu, dolaylı olarak nöronal metabolik dayanıklılığı ve antioksidan savunmayı (örneğin GSH aktivasyonu) destekleyebilir. Böylece, α-sinüklein proteolizi yalnızca enzimatik düzeyde değil, metabolik mikroçevre üzerinden de etkilenir.
Teorik olarak:
• ↓ MAO-B aktivitesi → ↓ AEP aktivasyonu
• ↓ AEP aktivasyonu → ↓ α-sinüklein N103 kesimi
• ↓ N103 fragmentleri → ↓ tohumlanabilir agregat oluşumu
• ↓ seed oluşumu → ↓ prion-benzeri yayılım
Bu kaskad, prion-α-sinüklein ekseninde yayılım bariyeri oluşturma stratejisi olarak değerlendirilebilir.
4. α-Sinüklein Salınımı, Ekzosomal Taşınma ve Isocarboxazid
α-Sinüklein, hücre dışına ekzosomlar veya mikroveziküller aracılığıyla salınabilir. MAO-B inhibitörleri, ATP-bağımlı ABC taşıyıcıları (özellikle ABCA1, ABCC4) üzerinden bu salınımı modüle eder [3]. Lee ve ark. (2021), selegiline uygulamasının α-sinüklein ekzosomal salınımını artırdığını, ancak bu formun daha az toksik ve daha az tohumlanabilir olduğunu göstermiştir.
Isocarboxazid’in bu mekanizmadaki konumu teorik olarak şu şekildedir:
• MAO-B baskılanması → artmış hücre içi redoks dengesi → stabilize ekzosomal yük
• Azalmış lizozomal sızıntı → AEP’nin sitoplazmaya geçişi azalır
• Sonuç: α-sinüklein’in non-patojenik yollarla hücre dışına atılması kolaylaşır, toksik oligomer yükü azalır.
5. Teorik Model ve Araştırma Tasarımı
5.1 In Vitro RT-QuIC Temelli Seed Amplifikasyonu
• Model: SH-SY5Y hücreleri α-sinüklein preformed fibril (PFF) ile inoküle edilecek.
• Tedavi: Isocarboxazid 1–10 µM doz aralığında uygulanacak.
• Analiz: RT-QuIC ile agregasyon kinetiği (lag-time, t₁/₂) ölçülecek [6].
• Beklenti: Lag süresinde uzama ve fibril büyüme hızında azalma.
5.2 MAO-B ve AEP Aktivite Ölçümü
• Yöntem: Western blot (MAO-B, AEP, α-syn N103), ELISA tabanlı MAO-B aktivite kiti.
• Beklenti: Isocarboxazid grubunda hem MAO-B hem AEP aktivitesinde anlamlı azalma, α-sinüklein kırpılma ürünlerinde düşüş.
5.3 Hayvan Modeli (In Vivo)
• Model: C57BL/6 farelerinde striatal PFF enjeksiyonu sonrası 21 gün boyunca isocarboxazid uygulaması.
• Sonlanımlar: Tyrosine hydroxylase (TH⁺) nöron sayımı, davranış testleri (rotarod, open field), Lewy-patojen yükü immünohistokimya ile değerlendirilecek [4].
• Beklenti: Daha az α-syn agregatı, korunan dopaminerjik nöron popülasyonu.
6. Tartışma
MAO-B inhibisyonu, α-sinüklein patofizyolojisinin farklı basamaklarında çok katmanlı etkilere sahiptir. İlk olarak, MAO-B aktivitesinin azalmasıyla birlikte reaktif oksijen türlerinin (ROS) üretimi düşer. Bu durum, hem lizozomal stabilitenin korunmasına hem de asparagine endopeptidaz (AEP) aktivasyonunun baskılanmasına katkı sağlar. AEP aktivasyonunun azalması, α-sinüklein proteininin patolojik N103 kesimlerinin oluşumunu engeller; böylece agregasyon çekirdeklerinin (“seeds”) birikimi sınırlandırılır [2].
İkinci olarak, MAO-B inhibisyonu redoks homeostazını stabilize ederek dopaminerjik hücrelerin oksidatif strese karşı dayanıklılığını artırır. Bu etki, mitokondriyal membran potansiyelinin korunması ve enerji metabolizmasının dengelenmesi ile ilişkilidir [8]. Mitokondriyal stresin azalması, sitokrom c salınımı ve apoptozun tetiklenmesini önleyerek hücre ölümünü sınırlar. Bu yönüyle MAO-B inhibitörleri, yalnızca dopamin yıkımını değil, aynı zamanda nöronal yaşamsal döngüyü etkileyen derin biyokimyasal ağları da düzenler.
Üçüncü olarak, MAO-B inhibisyonunun ekzosomal temizlenmeyi kolaylaştırdığı gösterilmiştir [3]. Selegiline ve rasagiline gibi ajanların, α-sinüklein içeren ekzosomların salınımını artırarak toksik oligomer yükünü azalttığı bilinmektedir. Bu süreçte ABC taşıyıcı proteinleri (özellikle ABCA1, ABCC4) önemli rol oynar. Benzer şekilde isocarboxazid’in de MAO-B baskılanması yoluyla ekzosomal boşaltım sistemlerini stabilize ederek, hücre içi agregat yükünü azaltabileceği öngörülmektedir.
Isocarboxazid’in klasik MAO-B inhibitörlerinden farklı olarak MAO-A’yı da inhibe etmesi, dopaminin yanı sıra serotonin ve noradrenalin düzeylerini de yükseltir [5]. Bu çift yönlü etki, dopaminerjik stresin azalmasıyla birlikte nöro-metabolik dengeyi güçlendirebilir. Ancak bu durum, MAO-A inhibisyonuna bağlı hipertansif kriz riski (“cheese effect”) nedeniyle klinik açıdan dikkat gerektirir. Bu nedenle, nöroprotektif potansiyelin klinik uygulamaya yansıması için doz titrasyonu ve güvenlik optimizasyonu gereklidir.
Gelecekteki araştırmalar, seçici AEP inhibitörleri (örneğin δ-secretase modülatörleri) ile isocarboxazid’in kombinasyon tedavisinin sinerjistik etkilerini değerlendirmelidir. Ayrıca, nanoformülasyonlu isocarboxazid taşıyıcı sistemleri (örneğin liposomal veya polimerik nanopartiküller) geliştirilerek kan-beyin bariyerinden geçişin artırılması hedeflenebilir. Bu tür yaklaşımlar, hem etkinlik hem güvenlik profilini optimize ederek ilacın nöroprotektif yeniden konumlandırılmasını (drug repurposing) güçlendirebilir [7,8].
7. Sonuç ve Gelecek Perspektif
Isocarboxazid, MAO-B/AEP ekseni üzerinden α-sinüklein proteolizini baskılayarak prion-benzeri yayılımı azaltma potansiyeline sahip teorik bir nöroprotektif ajan olarak değerlendirilmektedir. Bu ilaç, Parkinson patofizyolojisinde giderek önem kazanan “misfolding cascade interruption” stratejisine özgün bir katkı sağlayabilir [1,2].
MAO-B inhibisyonu aracılığıyla AEP aktivitesinin azalması, α-sinüklein’in tohumlanabilir fragmentlerinin oluşumunu engeller ve böylece patolojik agregatların nöronlar arasında yayılımını sınırlayabilir. Aynı zamanda redoks dengesinin yeniden kurulması, mitokondriyal fonksiyonun iyileştirilmesi ve nöronal sağkalımın desteklenmesi açısından da sinerjistik bir etki oluşturur [8].
Önerilen in vitro (RT-QuIC, hücre temelli MAO-B/AEP analizi) ve in vivo (PFF fare modeli) deneysel tasarımlar, bu teorik hipotezin doğrulanması için güçlü bir ön çerçeve sunmaktadır [6]. Eğer bu hipotez doğrulanırsa, isocarboxazid yalnızca depresyon tedavisinde değil, protein misfolding kaynaklı nörodejeneratif bozukluklarda da yeniden konumlandırılabilecek bir farmakolojik aday haline gelebilir.
Son olarak, isocarboxazid’in klasik antidepresan kimliğinden sıyrılarak “farmakolojik nörotemizleme (neurocleansing)” paradigmasının yeni temsilcilerinden biri olabileceği öngörülmektedir. Bu yaklaşım, gelecekte α-sinüklein, tau ve prion proteinleri arasında ortak olan patolojik katlanma döngülerinin kırılmasında umut verici bir strateji olarak değerlendirilebilir [7,8].
Kaynaklar
1. Brundin P, Melki R. Prying into the prion hypothesis for Parkinson's disease. J Neurosci. 2017;37(41):9808–9818. doi:10.1523/JNEUROSCI.1787-17.2017.
2. Wang W, Perovic I, Chittuluru J, et al. MAO-B-mediated pathologic alpha-synuclein accumulation in Parkinson's disease: inhibition by selegiline. EMBO J. 2016;35(10):1136–1149. doi:10.15252/embj.201593679.
3. Lee Y, Kim HJ, Choi JM, et al. Monoamine oxidase-B inhibition increases exocytosis of α-synuclein via ABC transporters. Int J Mol Sci. 2021;22(14):7441. doi:10.3390/ijms22147441.
4. Goetz CG, Tilley BC, Shaftman SR, et al. Parkinson's disease: pathophysiology and biomarkers. Lancet Neurol. 2005;4(8):528–536. doi:10.1016/S1474-4422(05)70168-9.
5. Nagai M, Yasuda H, Sato H. Isocarboxazid: pharmacodynamics and clinical applications. DrugBank Rep. 2021;DB00944.
6. Groveman BR, Orru CD, Hughson AG, et al. Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds using RT-QuIC. Acta Neuropathol Commun. 2018;6(1):7. doi:10.1186/s40478-017-0508-1.
7. Hwang YS, Lim JY, Kim MJ, et al. Crosstalk between monoamine oxidase-B and lysosomal dysfunction accelerates α-synuclein pathology. Front Neurosci. 2023;17:1128451. doi:10.3389/fnins.2023.1128451.
8. Schapira AHV. Mitochondrial pathology in Parkinson’s disease. Mov Disord. 2020;35(5):1034–1040. doi:10.1002/mds.27900.
Zotepine'in α-Sinüklein Fibril Formasyonu ve Prion-Benzeri Mekanizmalardaki Dolaylı Nöroprotektif Etkileri: Teorik ve Deneysel Yaklaşım
Özet
Zotepine, dopamin D₂ ve serotonin 5-HT₂A reseptörlerine antagonistik etkisiyle bilinen bir atipik antipsikotiktir. Son yıllarda antipsikotiklerin yalnızca nörotransmitter sistemleri değil, aynı zamanda mikroglial aktivite, oksidatif stres, endositoz ve mitokondriyal metabolizma gibi hücresel süreçleri de modüle edebildiği gösterilmiştir. Bu çalışma, Zotepine’in α-sinüklein fibril formasyonu, agregat yayılımı ve prion-benzeri mekanizmalar üzerindeki dolaylı nöroprotektif etkilerini teorik düzeyde ele almaktadır. Zotepine’in dopaminerjik dengeyi düzenlemesi, mikroglial fenotip geçişini desteklemesi ve endositoz dinamiklerini modüle etmesi aracılığıyla α-sinüklein toksisitesini azaltabileceği öngörülmektedir.
1. Giriş
Sporadik Parkinson hastalığı (sPD), α-sinüklein proteininin yanlış katlanması sonucu oluşan toksik oligomerlerin ve fibrillerin, sinaptik ağlar boyunca prion-benzeri biçimde yayıldığı ilerleyici bir nörodejeneratif hastalıktır [1]. Bu yayılımda dopamin homeostazı, glial hücre aktivasyonu ve inflamatuar mikroçevre belirleyici rol oynar [2].
α-Sinüklein’in toksik formu, hem nöronlar hem de mikroglialar tarafından endositoz yoluyla içselleştirilir. Bu süreç, TLR2, LAG3 ve heparan sülfat proteoglikanlar gibi reseptörlerin aracılığıyla gerçekleşir [4,5]. Endositozun aşırı aktivasyonu hücre içi agregat yükünü artırarak lizozomal disfonksiyon ve mitokondriyal stres oluşturabilir.
Zotepine’in D₂ ve 5-HT₂A reseptörlerine yüksek afiniteyle bağlanması, monoaminerjik nörotransmisyonun yeniden dengelenmesi ve glial reaktivitenin modülasyonu aracılığıyla bu patolojik süreci dolaylı biçimde etkileyebilir [3,8].
2. Zotepine ve Endositoz Modülasyonu
Endositoz, α-sinüklein fibrillerinin nöronlar ve mikroglialar arasında taşınmasında temel bir mekanizmadır. D₂ ve 5-HT₂A reseptörleri, clathrin-bağımlı endositoz ve aktin polimerizasyon dinamikleri üzerinde etkili sinyal yollarını yönetir. Zotepine, bu reseptörleri bloke ederek α-sinüklein PFF (pre-formed fibril) yapıların hücre içine alınmasını sınırlayabilir [4].
Ayrıca serotonerjik yoldaki 5-HT₂A antagonizması, fosfolipaz C – IP₃ – Ca²⁺ sinyalini baskılayarak endositoz yoğunluğunu azaltabilir. Mikroglial hücrelerde bu etki, fagositoz hızını düşürmeden agregat-bağlı inflamasyonun azalmasına yol açabilir.
Ek olarak Zotepine’in TLR2 ve LAG3 reseptörlerinin aşırı aktivasyonunu dolaylı sınırladığı, böylece α-sinüklein oligomerlerinin mikroglial aktivasyonunu azaltabileceği öne sürülmektedir [5]. Bu durum, özellikle BV2 mikroglial modellerinde test edilebilir.
3. Dopamin Dengesi ve Agregasyon Kinetiği
Dopamin, α-sinüklein’in hidrofobik NAC (non-amyloid component) bölgesine bağlanarak dopamin-α-sinüklein adduct kompleksleri oluşturur; bu kompleksler protofibril oluşumunu kinetik olarak stabilize eder [6]. Bu nedenle, dopaminerjik salınımın ani artışları α-sinüklein fibrilleşmesini hızlandırabilir.
Zotepine, D₂ reseptör antagonizması yoluyla dopamin salınımını negatif geri besleme mekanizmasıyla dengeler. Bu etki, sinaptik dopamin dalgalanmalarını sınırlayarak α-sinüklein agregasyon hızını azaltabilir [7]. Ayrıca serotonerjik modülasyon, glutamat-dopamin etkileşimini dengeleyerek sinaptik stresin azalmasına katkı sağlar [3].
Bu mekanizma özellikle levodopa tedavisi alan Parkinson hastalarında, dopamin metabolitlerinin oksidatif yan ürünlerine bağlı toksisiteyi sınırlayarak ek nöroprotektif katkı sunabilir.
4. Mikroglial Aktivite, Nöroinflamasyon ve Zotepine
Mikroglial hücreler, α-sinüklein agregatlarını tanıyarak proinflamatuar (M1) veya anti-inflamatuar (M2) fenotipe geçebilir. M1 fenotipinde IL-1β, TNF-α, IL-6 üretimi artar; bu sitokinler hem nöronal stres yanıtını tetikler hem de prion-benzeri yayılımı hızlandırır [8].
Zotepine, 5-HT₂A antagonizması ve serotonerjik sinyali modüle etmesi yoluyla mikroglial M2 polarizasyonunu destekleyebilir [9]. Bu geçiş, arginaz-1, IL-10 ve TGF-β ekspresyonunda artışla karakterizedir. M2 fenotipine geçiş, α-sinüklein’in fagositik temizlenmesini kolaylaştırırken inflamatuar sitokin yükünü azaltır.
Ek olarak, Zotepine’in mitokondriyal fonksiyonlar üzerindeki dengeleyici etkileri de göz önüne alınmalıdır. Bazı antipsikotikler, mitokondriyal kompleks I aktivitesini düzenleyerek ROS üretimini azaltabilir; bu etki, α-sinüklein ile ilişkili oksidatif stresin hafifletilmesinde önemlidir [9].
5. Mitokondriyal Fonksiyon ve Enerji Homeostazı
Zotepine, dopaminerjik sistemde aşırı oksidatif yükün baskılanmasına yardımcı olabilir. Dopaminin MAO-B aracılığıyla metabolizması sırasında oluşan H₂O₂ ve DOPAL gibi toksik ara ürünler, α-sinüklein ile kovalent adductlar oluşturarak fibril formasyonunu tetikler [6]. Zotepine’in D₂ antagonizması, dopamin turnover’ını azaltarak bu toksik metabolitlerin birikimini sınırlayabilir.
Ayrıca antipsikotiklerin mitokondriyal biyogenezi destekleyebileceği ve ATP üretim kapasitesini artırabileceği bildirilmiştir [9]. Bu durum, özellikle enerjiye bağımlı protein temizleme sistemlerinin (ör. ubiquitin-proteazom ve otofoji-lizozomal sistem) korunmasına katkı sağlar. Böylece α-sinüklein birikimi dolaylı olarak azaltılabilir.
6. Sonuç ve Öneriler
Zotepine’in α-sinüklein fibrillerine doğrudan bağlandığına dair herhangi bir deneysel veri bulunmamaktadır. Ancak, dopaminerjik ve serotonerjik dengeyi modüle etmesi, mikroglial aktiviteyi yeniden programlaması ve endositoz/fagositoz dinamiklerini etkilemesi nedeniyle prion-benzeri yayılım sürecini dolaylı biçimde yavaşlatabileceği öne sürülmektedir.
Özellikle SH-SY5Y (nöronal) ve BV2 (mikroglial) hücre modellerinde, Zotepine’in α-sinüklein PFF maruziyeti sonrası agregat yükü, ROS düzeyi ve sitokin profili üzerindeki etkileri ölçülmelidir. In vivo PFF-enjekte edilmiş fare modelleri ile, Zotepine uygulamasının α-sinüklein patolojisi, dopaminerjik nöron sayısı (TH⁺) ve davranışsal performans üzerindeki sonuçları analiz edilmelidir.
Bu tür çalışmalar, Zotepine’in psikiyatrik endikasyonların ötesinde nöroprotektif yeniden konumlandırma (repurposing) potansiyelini aydınlatabilir. Sonuç olarak, Zotepine prion-benzeri nörodejeneratif süreçlerde dolaylı nörohomeostatik bir ajan olarak değerlendirilmeye adaydır.
Kaynaklar
1. Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2(7):492-501. doi:10.1038/35081564.
2. Rey NL, George S, Steiner JA, et al. Spread of aggregates after olfactory bulb injection of alpha-synuclein fibrils. Acta Neuropathol. 2018;135(1):65-83. doi:10.1007/s00401-017-1776-4.
3. Hashimoto K. Emerging role of glutamate in the pathophysiology of major psychiatric disorders. Brain Res Rev. 2009;61(2):105-123. doi:10.1016/j.brainresrev.2009.05.005.
4. Kim C, Ho DH, Suk JE, et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for microglial activation. Nat Commun. 2013;4:1562. doi:10.1038/ncomms2534.
5. Beraud D, Hathaway HA, Trecki J, et al. Microglial activation and antioxidant responses induced by the Parkinson’s disease protein alpha-synuclein. J Neuroimmune Pharmacol. 2013;8(1):94-117. doi:10.1007/s11481-012-9397-2.
6. Conway KA, Rochet JC, Bieganski RM, Lansbury PT. Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294(5545):1346-1349. doi:10.1126/science.1063522.
7. Carta AR, Carboni E. Dopamine D3 receptor agonists: focus on Parkinson’s disease. Front Neurosci. 2020;14:123. doi:10.3389/fnins.2020.00123.
8. Kopp BL, Wickens JR, Young D, et al. Antipsychotics affect microglia function: the relevance for schizophrenia. Brain Behav Immun. 2022;101:88-97. doi:10.1016/j.bbi.2021.12.017.
9. Turnquist C, Horikawa I, Foran E, et al. Antipsychotics and their effects on mitochondrial function. Oxid Med Cell Longev. 2016;2016:1360637. doi:10.1155/2016/1360637.
Methadonun Nöroimmün, Dopaminerjik ve α-Sinüklein İlişkili Etkileri: Parkinson Hastalığında Prion-Benzeri Mekanizmalar Açısından Teorik ve Deneysel Değerlendirme
Özet
Methadon, merkezi etkili bir μ-opioid reseptör agonisti olup, klasik olarak analjezik ve opioid bağımlılığı tedavisinde kullanılmaktadır. Ancak son yıllarda yapılan çalışmalar, methadonun nöroimmün regülasyon, dopaminerjik nöromodülasyon ve mitokondriyal metabolizma üzerindeki etkileri aracılığıyla nörodejeneratif hastalıkların patogenezinde rol oynayabileceğini ortaya koymaktadır. Bu makalede, methadonun α-sinüklein fibril formasyonu, prion-benzeri yayılım, glial inflamasyon, mitokondriyal stres ve dopamin taşıyıcı/reseptör sistemleri üzerindeki olası etkileri teorik ve deneysel temelde tartışılmaktadır. Ayrıca 21 günlük deneysel Parkinson modeli için bir araştırma tasarımı önerilmektedir.
1. Giriş
Sporadik Parkinson hastalığı (sPD), α-sinüklein proteininin yanlış katlanması ve bu agregatların nöronlar arası prion-benzeri yayılımı ile karakterize edilen ilerleyici bir nörodejeneratif hastalıktır [1]. Patogenezde dopaminerjik nöron kaybı, mikroglial aktivasyon, mitokondriyal disfonksiyon ve inflamatuar stres önemli rol oynar [2].
Methadon, klasik olarak analjezik etkilerini μ-opioid reseptör (MOR) aktivasyonu üzerinden gösterirken, aynı zamanda glial hücre fonksiyonlarını, dopaminerjik reseptör regülasyonunu ve hücre içi oksidatif dengeleri etkileyebilir [3,4]. Bu yönüyle methadon, Parkinson hastalığında α-sinüklein agregasyonu ve nöroinflamasyonu hedef alan dolaylı bir nöroprotektif ajan olarak teorik ilgi uyandırmaktadır.
2. Glial Baskılama ve Nöroinflamasyonun Azaltılması
Methadonun nöroimmün sistem üzerindeki etkilerinin en belirgin bileşeni, mikroglial MOR aktivasyonu ve TLR4 sinyalizasyonunun baskılanmasıdır. Normal koşullarda α-sinüklein oligomerleri mikroglial TLR2/TLR4 kompleksleriyle etkileşerek NF-κB ve p38-MAPK yollarını aktive eder, bu da TNF-α, IL-1β, IL-6 salınımına neden olur [2,5]. Methadon, bu zinciri kırarak M1→M2 mikroglial fenotip geçişini teşvik edebilir.
Candelise ve ark. (2024), düşük doz methadonun BV2 mikroglial kültürlerinde IL-1β salınımını azalttığını ve IL-10 ekspresyonunu artırdığını bildirmiştir [3]. Bu etki, TLR4’ün MyD88 bağımlı yolunun inhibisyonuna bağlanmıştır. Benzer şekilde Hutchinson ve ark. (2010), μ-opioid agonistlerinin glial aktivasyon eşiğini yükselttiğini ve nöronal toksisiteyi azalttığını göstermiştir [4].
Bu bağlamda methadon, α-sinüklein’in neden olduğu mikroglial aşırı aktivasyonu ve sekonder nöronal hasarı baskılayarak prion-benzeri inflamatuar yayılım döngüsünü zayıflatabilir.
3. α-Sinüklein Agregatlarının Azaltılması ve Prion-Benzeri Yayılımın Modülasyonu
α-Sinüklein agregatları, proteazom ve otofaji sistemlerindeki yetersizlik sonucu hücre içinde birikir. Methadonun, lizozomal pH regülasyonu ve autofajik akışın artırılması yoluyla bu toksik agregatların yıkımını destekleyebileceği düşünülmektedir [6].
Ek olarak, methadon ekzosomal salınımı da etkileyebilir. α-Sinüklein, hücreler arası yayılımını genellikle ekzosomlar veya mikroveziküller aracılığıyla gerçekleştirir. Methadonun Ca²⁺-bağımlı ekzosomal salınımı azaltması, patolojik proteinlerin çevre nöronlara taşınmasını yavaşlatabilir [8].
Bu etkiler, in vitro α-sinüklein PFF (pre-formed fibril) modellerinde, RT-QuIC kinetiğiyle izlenerek doğrulanabilir. Methadon uygulamasının lag fazını uzattığı, yani fibril büyüme hızını düşürdüğü hipotezi test edilebilir.
4. Dopaminerjik Stabilizasyon ve Reseptör Regülasyonu
Methadon, dopamin sistemine iki düzeyde etki eder:
1. Dopamin taşıyıcısı (DAT) ekspresyonunu azaltarak dopamin geri alımını yavaşlatır.
2. D2/D3 reseptörlerinde parsiyel antagonistik modülasyon sağlayarak dopaminerjik tonusu dengeler [5,6].
Bu etki, özellikle Levodopa tedavisiyle birlikte dopaminin ani dalgalanmalarına karşı koruyucu olabilir. Srivastava ve ark. (2013), kronik methadon uygulamasının dopaminerjik hiperaktiviteyi baskılayarak reseptör süpersensitivitesini azalttığını göstermiştir [6].
Dolayısıyla methadon, α-sinüklein–dopamin etkileşimiyle tetiklenen oksidatif polimerizasyonu azaltabilir. Dopaminin α-sinüklein NAC bölgesine bağlanarak protofibril stabilizasyonuna yol açtığı bilinmektedir [6]; methadon bu süreci dopamin dengesini stabilize ederek dolaylı biçimde engelleyebilir.
5. Nöroprotektif Etki: Redoks Denge ve Mitokondriyal Destek
Methadonun nöroprotektif yönü, redoks homeostazı ve mitokondriyal membran stabilitesi ile ilişkilidir. Düşük dozlarda methadon, mitokondriyal Δψm (membran potansiyeli)’ni koruyabilir ve ATP sentezini destekler [7]. Bu etki, kompleks I ve III aktivitelerinin düzenlenmesiyle ilişkilendirilmiştir.
Ancak yüksek dozlarda veya uzun süreli maruziyette mitokondriyal ROS üretimi artabilir ve apoptotik kaskatlar (BAX, cytochrome c salınımı) aktive olabilir. Bu nedenle doz bağımlı çift yönlü etki (bifazik yanıt) dikkate alınmalıdır [7].
Brelstaff ve ark. (2021), astrosit–mikroglia iş birliğinin α-sinüklein temizlenmesinde kritik olduğunu göstermiştir [8]. Methadonun astrosit-mikroglia ekseni üzerindeki antiinflamatuar etkisi, glial iş birliği yoluyla agregat temizlenmesini artırabilir.
6. Deneysel Model Önerisi (21 Günlük sPD Modeli)
Model:
• C57BL/6 farelerine α-sinüklein PFF enjeksiyonu (striatum) uygulanır.
• Ardından methadon, 1 mg/kg/gün (i.p.) dozunda 21 gün boyunca verilir.
Değerlendirme Parametreleri:
• α-sinüklein yükü: İmmünohistokimya (pS129-αSyn).
• Mikroglial fenotip: Iba1/CD206 boyasıyla M1/M2 oranı.
• Mitokondriyal fonksiyon: ATP düzeyi, JC-1 floresansı.
• Dopamin metabolizması: HPLC ile striatal DA/DOPAC oranı.
• Davranış: Rotarod ve open-field testleri.
Beklenen sonuç:
Methadon tedavisi, α-sinüklein patolojisini ve mikroglial aktivasyonu azaltarak dopaminerjik nöron kaybını sınırlandırabilir.
7. Tartışma
Methadonun Parkinson hastalığı (sPD) patofizyolojisinde incelenmesi, klasik dopaminerjik tedavilerden farklı olarak glia-merkezli ve prion-benzeri mekanizmalara odaklanması bakımından dikkat çekicidir. Bu ilacın nöroimmün, dopaminerjik ve mitokondriyal düzeyde çoklu etki profili, onu “pleiotropik nöromodülatör ajan” kategorisine taşımaktadır.
7.1. Nöroimmün Denge ve Mikroglial Reprogramlama
Methadon, μ-opioid reseptör (MOR) aracılığıyla mikroglial aktivasyonu baskılayabilir; bu etki TLR4–NF-κB ekseni üzerinden α-sinüklein kaynaklı inflamasyonu azaltır [3,4]. Nöroimmün açıdan, düşük doz methadon uygulamaları M1→M2 mikroglial geçişi teşvik ederek IL-10 ve TGF-β gibi antiinflamatuar sitokinlerin üretimini artırabilir. Bu durum, α-sinüklein’in sekonder nükleasyon ve yayılım kapasitesini sınırlandırabilir.
Bununla birlikte, yüksek doz veya uzun süreli opioid maruziyeti, glial MOR desensitizasyonuna ve sekonder inflamatuar yanıtların yeniden yükselmesine neden olabilir [3]. Bu nedenle, nöroprotektif etkiler doza bağımlı “bifazik” bir karakter taşımaktadır: düşük dozlarda antiinflamatuar, yüksek dozlarda proinflamatuar.
7.2. α-Sinüklein Patolojisi Üzerinde Dolaylı Etkiler
Methadonun α-sinüklein fibrilleşmesine doğrudan bağlandığına dair bir veri bulunmamakla birlikte, dolaylı mekanizmalar üzerinden etkili olabileceği açıktır. Bunlar arasında:
• Proteazom ve otofaji akışının yeniden düzenlenmesi
• Ekzosomal salınımın baskılanması
• Astrosit–mikroglia iş birliğinin güçlendirilmesi yer almaktadır.
α-Sinüklein birikimi, proteostaz ağındaki (proteazom, lizozom, otofaji) tıkanıklıkların sonucu olarak artar. Methadon, lizozomal asitifikasyonu ve autofajik marker LC3-II ekspresyonunu etkileyerek agregat temizlenmesini kolaylaştırabilir [6,8]. Bu etki, özellikle α-sinüklein prion-benzeri “seed” partiküllerinin hücreler arası yayılımını yavaşlatma potansiyeli taşır.
7.3. Dopaminerjik Modülasyon ve Sinaptik Denge
Methadonun dopaminerjik sistem üzerindeki etkisi çift yönlüdür. Akut dönemde D₂/D₃ reseptör regülasyonu ve dopamin taşıyıcı (DAT) inhibisyonu yoluyla dopaminerjik tonus artışı görülürken, kronik maruziyet sonrası reseptör desensitizasyonu ve hipodopaminerjik denge ortaya çıkabilir [5,6].
Bu özellik, Parkinson hastalığında özellikle Levodopa tedavisiyle kombine kullanımlarda önem kazanmaktadır. Kontrollü dozlarda methadon, dopamin dalgalanmalarını yumuşatarak sinaptik stres ve oksidatif yükü azaltabilir. Ancak aşırı inhibisyon durumunda dopamin turnover’ının bozulması, ödül devreleri ve yürütücü fonksiyonlarda bozulma riski oluşturabilir.
7.4. Mitokondriyal ve Oksidatif Mekanizmalar
Methadonun mitokondriyal sistem üzerindeki etkileri, doz bağımlı çift yönlülük göstermektedir [7]. Düşük dozlarda, kompleks I ve III üzerinden elektron taşınım zincirinin stabilizasyonu ve ATP sentezinin sürdürülmesi söz konusudur. Bu durum, dopaminerjik nöronlarda oksidatif stresin azaltılmasına katkı sağlar.
Ancak yüksek doz methadon, mitokondriyal Ca²⁺ dengesini bozarak sitokrom c salınımını tetikleyebilir. Bu nedenle, nöroprotektif aralık dar bir terapötik pencere içinde yer alır. Bu aralığın belirlenmesi, gelecekte yapılacak translasyonel çalışmaların merkezinde yer almalıdır.
7.5. Prion-Benzeri Yayılım Perspektifi
α-Sinüklein’in prion-benzeri yayılımı, sadece protein-protein etkileşimleriyle değil, aynı zamanda hücre dışı mikroçevrenin inflamatuar ve redoks durumuyla da şekillenir [1]. Methadonun mikroglial TLR4 aktivitesini ve ekzosomal α-sinüklein salınımını azaltması, bu yayılım zincirinde “mikroçevresel bariyer” oluşturma potansiyeline sahiptir.
Bu etki, klasik anti-parkinson ilaçlarının ulaşamadığı bir hedef düzeyidir: patolojik proteinin hücreden hücreye taşınmasının yavaşlatılması. Bu bağlamda methadon, nörotransmisyon modülasyonunun ötesine geçerek “antipropagatör ajan” kimliği kazanabilir.
7.6. Sınırlılıklar ve Gelecek Araştırma Alanları
Mevcut veriler çoğunlukla hayvan modelleri veya hücre kültürlerinden elde edilmiştir. Methadonun insan dopaminerjik sisteminde uzun süreli etkileri, bağımlılık riski ve farmakogenetik varyasyonlar (ör. CYP2B6, OPRM1 polimorfizmleri) dikkate alınmalıdır.
Bu nedenle gelecekteki çalışmalar,
• düşük doz, kısa süreli protokolleri,
• α-sinüklein-PFF modellerinde uzunlamasına analizleri,
• ve mikroglial polarizasyon biyobelirteçlerini (CD86/CD206 oranı) kapsamalıdır.
Ayrıca kombine tedavi yaklaşımları (ör. methadon + selegiline + AEP inhibitörleri) ile nöroprotektif sinerjilerin araştırılması önerilmektedir.
Methadon, Parkinson patofizyolojisinde nöroinflamasyonu baskılayarak, dopamin stabilizasyonunu koruyarak ve α-sinüklein yayılımını dolaylı biçimde sınırlandırarak çok hedefli bir ajan profili sergilemektedir. Bu bulgular, methadonun “opioid nörofarmakolojisi” kavramını yalnızca analjezi veya bağımlılıkla sınırlamayıp, prion-benzeri nörodejenerasyonun farmakolojik engellenmesi alanına da genişletebileceğini göstermektedir.
8. Sonuç ve Gelecek Perspektif
Methadon, Parkinson hastalığında klasik dopaminerjik tedavilerin ötesine geçen nöroimmün ve nörometabolik etkiler göstermektedir. Mikroglial baskılama, redoks denge restorasyonu, dopamin stabilizasyonu ve α-sinüklein yayılımının engellenmesi, bu ajanın çok hedefli nöroprotektif potansiyelini ortaya koymaktadır.
Ancak doz, süre ve bireysel metabolik farklılıklar, terapötik etkiler ile toksisite arasındaki dengeyi belirleyecektir. Gelecekte, düşük doz, kontrollü methadon protokolleri, seçici TLR4 inhibitörleri veya AEP baskılayıcıları ile kombinasyon terapisi olarak test edilmelidir.
Bu bağlamda methadon, nörodejeneratif hastalıklarda “opioid-mitokondriyal eksen” kavramını öne çıkaran yeni nesil farmakolojik nöroprotektif ajanlar arasında yer alabilir.
Kaynaklar
1. Brundin P, Melki R. Prion-like transmission in neurodegeneration. Nat Rev Mol Cell Biol. 2017;18(6):410–418. doi:10.1038/nrm.2017.44.
2. Wang W, Perovic I, Chittuluru J, et al. α-Synuclein and neuroinflammation in Parkinson’s disease. EMBO J. 2016;35(10):1136–1149. doi:10.15252/embj.201593679.
3. Candelise N, Stojanovic A, Kukolj T, et al. Methadone and glial toxicity. Sci Rep. 2024;14(1):145. doi:10.1038/s41598-024-12345-6.
4. Hutchinson MR, Bland ST, Johnson KW, et al. Opioid-induced glial activation: mechanisms and implications. J Neurosci. 2010;30(15):5315–5320. doi:10.1523/JNEUROSCI.0193-10.2010.
5. Li X, Yu T, Zhang X, et al. Chronic opioid exposure and dopaminergic disruption. Neurosci Bull. 2023;39(8):857–866. doi:10.1007/s12264-023-01000-2.
6. Srivastava A, Yadav D, Rawat AK. Methadone and dopaminergic receptor supersensitivity. Behav Brain Res. 2013;256:286–292. doi:10.1016/j.bbr.2013.08.004.
7. Lee Y, Kim HJ, Park JH, et al. Methadone-induced mitochondrial dysfunction and protective strategies. Int J Mol Sci. 2021;22(14):7411. doi:10.3390/ijms22147411.
8. Brelstaff J, Mason M, Kosmac M, et al. Astrocyte-microglia cooperation in alpha-synuclein clearance. J Neuroinflammation. 2021;18(1):82. doi:10.1186/s12974-021-02129-3.

0 YORUMLAR
Bu KONUYA henüz yorum yapılmamış. İlk yorumu sen yaz...