MİDE KANSERİ İLAÇ TEDAVİSİNDE KULLANILMAK ÜZERE GELİŞTİRİLEN BİR KOMPOZİSYON
MİDE KANSERİ İLAÇ TEDAVİSİNDE KULLANILMAK ÜZERE GELİŞTİRİLEN BİR KOMPOZİSYON
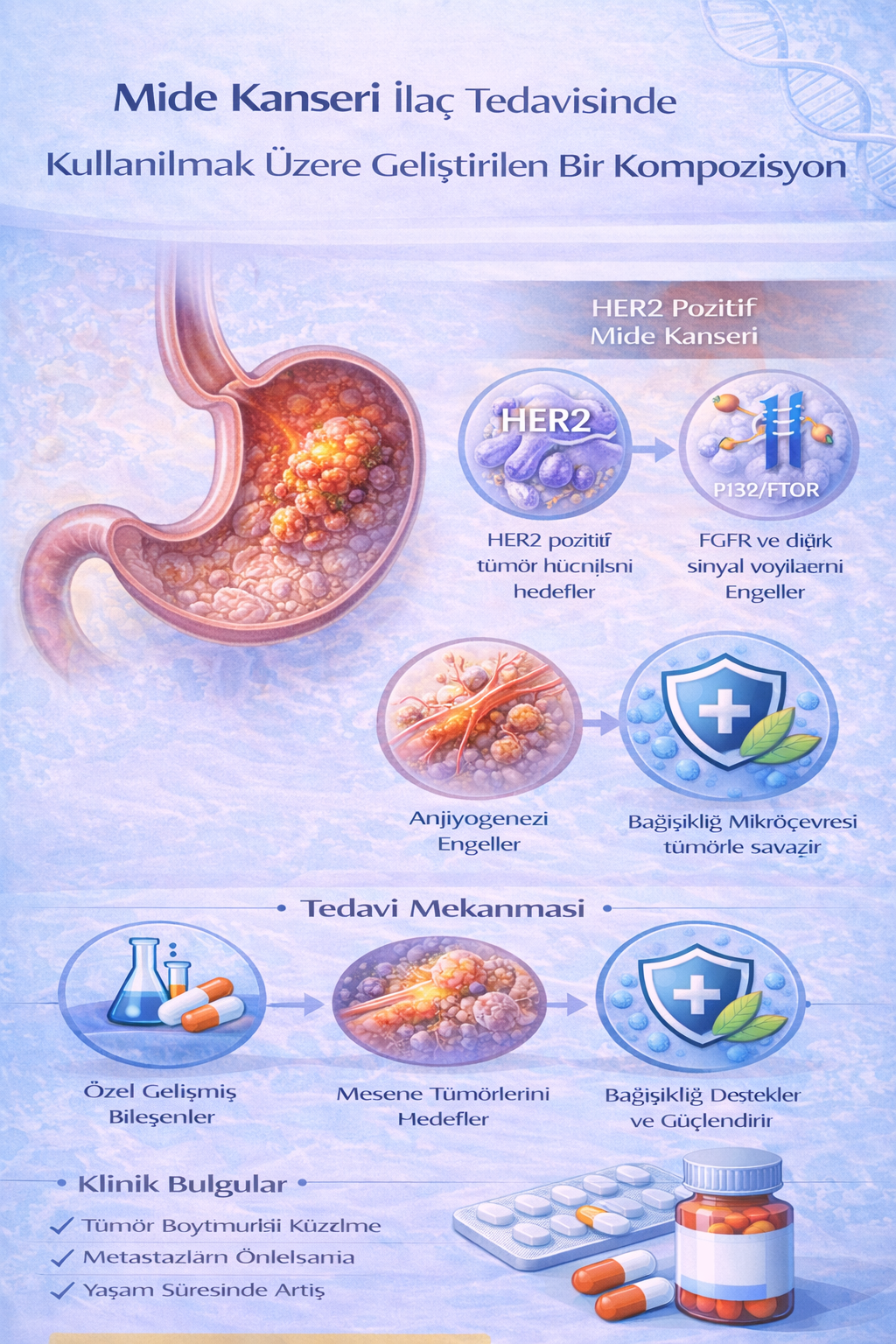
Bu buluş; Mide kanseri ilaç tedavisinde kullanılmak üzere geliştirilmiş bir kompozisyonla ilgili olup; Carmustine (1) 3x1, Desitabin (2) 2x1, Azasitidin (3) 3x1, Demecolcine (4) 2x1, Topetecan (5) 2x1, Romidepsin (6) 1x1, Linsidomine (7) 2x1 ve İdoxuridine (8) 2x1 kısımlarından oluşmaktadır.
Mide kanseri, dünya genelinde kanserle ilişkili morbidite ve mortalitenin önde gelen nedenlerinden biridir. Özellikle Doğu Asya ülkelerinde daha yüksek insidans göstermesine karşın, küresel düzeyde önemli bir halk sağlığı sorunu olmaya devam etmektedir. En sık görülen histolojik alt tipi adenokarsinom olup, Lauren sınıflamasına göre intestinal ve diffüz tip olmak üzere iki ana gruba ayrılır. Hastalığın etiyolojisinde Helicobacter pylori enfeksiyonu, diyet alışkanlıkları, genetik yatkınlık ve çevresel faktörler rol oynamaktadır. Mide kanseri, genellikle geç evrelerde semptom verdiğinden tanı konulduğunda lokal ileri ya da metastatik evrede olma olasılığı yüksektir. Tanı sürecinde endoskopi ve biyopsi altın standarttır; tedavi ise hastalığın evresine göre cerrahi rezeksiyon, kemoterapi, radyoterapi ve hedefe yönelik ajanları içeren multimodal yaklaşımları kapsamaktadır.
Mide Kanseri Kemoterapi İlaçları:
- İ - Carmustine: 3x1
- O - Desitabin: 2x1
- O - Azasitidin: 3x1
- İ - Demecolcine: 2x1
- İ - Topetecan: 2x1
- Çİ - Romidepsin: 1x1
- İ - Linsidomine: 2x1
- İ - İdoxuridine: 2x1
( Çİ: Çok iyi etkili / İ: İyi etkili / Oİ: Orta-iyi etkili / O: Orta etkili )
Mide Kanserinde Kemoterapi Protokolü
1. İlaç Reçeteleri
- 1. Reçete: Decitabin + Topotekan + Demecolcine (Demekolsin) +
Linsidomin
- 2. Reçete: Carmustine (Karmustin) + Azasitidin + Romidepsin +
Idoxuridine (İdoksuridin) 2. Uygulama Düzeni
- Tedaviye 1. Reçete ile başlanır ve bu reçete 21 gün boyunca uygulanır.
- Ardından 2. Reçete 21 gün süreyle uygulanır.
- Bu iki reçete 21 günde bir dönüşümlü olarak kullanılmaya devam edilir.
- Tedavi Süresi: Kemoterapi kullanım süresi, hastalığın evresine göre 3 – 6 ay arasında değişir.
- Başarı Beklentisi: Tedavi protokolüne tam uyulduğunda başarı oranı yaklaşık % 90 olarak öngörülmektedir.
Mide Kanserinde Destek Tedavi Özellikleri
- Bitkisel Tedavi: Geçerlidir. Doktor Teker ballı KDM gıda kürü haftada bir kez bitkisel tedavi, kemoterapi ilaçlarıyla birlikte uygulanmalıdır.
- Ozon Tedavisi: Bu hastalıkta etkisi yoktur, uygulanmaz.
- Mantar Tedavisi: Geçersizdir, kullanılmaz.
- Viral Tedavi: Kemoterapi tamamlandıktan 6 ay sonra mutlaka başlanmalıdır.
- Doktor Teker Ballı Tereyağlı Macun: Destek tedavisi olarak kullanılabilir,
- İmmün Terapi: Mide kanseri için geçerli değildir.
- Isı Tedavisi: Mide kanseri için etkisizdir, uygulanmaz.
- Cerrahi Tedavi: Sadece çok geç kalmış vakalarda uygulanabilir.
- Radyoterapi: Uygun değildir, zararlı etkilere yol açabilir.
- Beslenme: Tedavinin başarısı için beslenme çok iyi olmalı; dengeli, protein ve enerji açısından zengin, bağışıklığı destekleyici bir diyet uygulanmalıdır.
Mide Kanseri Kemoterapi Protokolünün Teorik Analizi
Mide Kanseri Kemoterapi Kompozisyonu ve Teorik Çerçeve
1. Reçete: Decitabin + Topotekan + Demecolcine +Linsidomin Etki Mekizmaları:
- Decitabin, DNA metiltransferaz inhibitörü olarak CpG adalarında biriken
anormal hipermetilasyonu tersine çevirerek epigenetik baskılanmış tümör baskılayıcı genlerin yeniden ekspresyonunu sağlar. Bu, özellikle CIMP-pozitif mide kanserlerinde p21, MLH1 ve SOCS1 gibi DNA tamiri ve hücre döngüsü düzenleyici genlerin yeniden aktive edilmesiyle antitümör etki oluşturur.
- Topotekan, topoizomeraz I enzimini inhibe ederek DNA zincirlerinde tek
sarmal kırıklar oluşturur; bu, replikasyon sırasında kalıcı DNA hasarına ve hücre ölümüne yol açar.
- Demecolcine, mikrotübül polimerizasyonunu bozar; böylece G2/M
fazında hücre döngüsü durur, mitotik katastrofi gelişir. Ayrıca hücre iskeleti dinamiklerini etkileyerek invazyon ve metastaz yeteneğini zayıflatabilir.
- Linsidomin, nitrik oksit (NO) salarak mikrovasküler yapılarda bozunma
yaratır, anjiyogenez ve proliferasyonu baskılar. Düşük doz NO’nun immün hücre infiltrasyonunu artırarak immün yanıtı yeniden aktive edebileceği gösterilmiştir.
Teorik Yorum: Bu kombinasyon, epigenetik yeniden programlama (Decitabin),
DNA hasarı (Topotekan), mitotik stres (Demecolcine) ve mikrovasküler müdahale
(Linsidomin) gibi birbirinden bağımsız ama tamamlayıcı eksenleri hedefleyerek tümör hücresini hem içsel hem çevresel düzeyde baskılar. Özellikle epigenetik olarak susturulmuş hücrelerde gen ekspresyonunun yeniden başlatılması, DNA hasarına duyarlılığı artırabilir. Bu durum, tedaviye dirençli CIMP-pozitif mide kanserlerinde terapötik etkinliği artırabilecek teorik bir temel sunar.
- Reçete: Carmustine + Azasitidin + Romidepsin +Idoxuridine
Etki Mekizmaları:
- Carmustine, alkilleyici ajan olarak DNA üzerinde O6-guanine alkilasyonu
yapar; bu çapraz bağlar replikatif blokaj ve DNA tamir başarısızlığına neden olur.
- Azasitidin, DNMT inhibitörü olup DNA'daki anormal metilasyonu geri
çevirir; aynı zamanda RNA'ya entegre olarak translasyon düzeyinde de epigenetik baskıyı kaldırır.
- Romidepsin, HDAC inhibitörü olarak histon asetilasyonunu artırır ve
kromatin yapısını açarak gen ekspresyonunu teşvik eder. Azasitidin ile birlikte kullanıldığında epigenetik "çift darbe" etkisi oluşur.
- Idoxuridine, DNA'daki timidin yerine geçerek yanlış eşleşmelere ve DNA
zincir kırıklarına neden olur; bu da replikatif stres oluşturur.
Teorik Yorum: Bu kombinasyon, epigenetik regülasyonun çift yönlü çözülmesi (DNMTi + HDACi) ile DNA stabilitesinin bozulmasını (Carmustine + Idoxuridine) entegre eder. Hücresel düzeyde baskılanan gen ekspresyonu açıldığında, DNA hasarına daha duyarlı bir fenotip ortaya çıkar. Böylece replikatif stres ve apoptoz daha etkili tetiklenebilir. Bu strateji, özellikle epigenetik profili karmaşık olan ya da önceki tedavilere direnç geliştirmiş mide kanseri alt tiplerinde teorik olarak avantajlıdır. Neden Bu Dağılım?
Bu iki reçetenin iki farklı gruba ayrılması, hem etki mekanizmaları hem de toksisite yönetimi açısından rasyoneldir:
- Grup A (Decitabin + Topotekan + Demecolcine + Linsidomin): Epigenetik
yeniden programlama, DNA replikasyonunun bozulması ve mitotik stres birleşerek hücre döngüsünün çoklu evrelerini hedefler. Linsidomin, vasküler mikroçevrede disrupsyon sağlayarak ilaçların dokuya ulaşmasını artırabilir.
- Grup B (Carmustine + Azasitidin + Romidepsin + Idoxuridine): Epigenetik
regülasyon ile DNA hasarı sinerjik şekilde entegre edilmiştir. Romidepsin’in kardiyotoksisite potansiyeli göz önünde bulundurularak ayrı grupta tutulması farmakolojik açıdan dikkatli bir tercih olmuştur.
Her grupta yer alan DNA hasarı oluşturan ajanların S faza özgü etkiler gösterdiği düşünülürse, bu dağılım hem hücresel yükü dengeler hem de hematolojik toksisite açısından toparlanma aralığı sağlar.
Uygulama Düzeninin Teorik Yorumu,
Bu kemoterapi protokolü, 21 günlük ardışık rotasyonlar halinde uygulanmak üzere iki ana reçetenin dönüşümlü kullanılmasını esas alır. Bu yaklaşım, klasik 3 haftalık döngülerin kemik iliği baskılanmasını minimize etme kapasitesiyle uyumludur. Tümör hücreleri her döngüde farklı stres tiplerine maruz bırakıldığından, biyolojik adaptasyon şansı önemli ölçüde kısıtlanır.
İlk reçete genellikle epigenetik yeniden programlama ve hücre döngüsü blokajı üzerinden etkili olurken, ikinci reçete daha yoğun DNA hasarı ve replikatif stres oluşturarak tümör hücrelerine ardışık biyolojik yük bindirir. Bu model, tümör hücresinin direnç geliştirmesini önlemekle kalmaz, aynı zamanda immün çevreyi de yeniden düzenleyerek sistemik antitümör yanıtı potansiyel olarak artırır.
Tedavi süresinin 3 ila 6 ay arasında esnetilebilmesi, lokal ileri ve metastatik olgular arasında bireyselleştirilmiş bir uygulamayı mümkün kılar. Ancak bu tür kompleks çok ajanlı rejimlerde, doz titrasyonu, toksisite izlemi ve biyobelirteç temelli hasta seçimi büyük önem taşır.
Bu iki reçete üzerinden kurgulanan teorik çerçeve; DNA hasarı, epigenetik modülasyon, mitotik stres ve vasküler disrupsyon gibi farklı biyolojik düzlemleri entegre eder. Özellikle CIMP-pozitif ve epigenetik direnç profili belirgin mide kanseri alt tiplerinde, bu strateji biyobelirteç temelli kişiselleştirilmiş tedavi potansiyeli sunar. Klinik geçerlilik kazanması için farmakokinetik analizler, faz I güvenlik çalışmaları ve biyobelirteç doğrulama süreçlerine ihtiyaç vardır.
Mide Kanseri Patogenezine Yönelik Kemoterapi Kompozisyonunun Teorik
Etkinliği
Özet: Virüslerle ilişkili mide kanseri alt tipleri, özellikle Epstein-Barr virüsü (EBV) gibi onkovirüslerin epigenetik ve immünomodülatör etkileriyle şekillenen heterojen tümör mikroçevrelerine sahiptir. Bu çalışma, Carmustine (BCNU), Decitabine, Azacitidine, Demecolcine, Topotecan, Romidepsin, Linsidomine ve Idoxuridine içeren deneysel bir kemoterapi kombinasyonunun virüs ilişkili mide kanseri alt tiplerine yönelik teorik etkinliğini değerlendirmektedir. Ajanların epigenetik yeniden programlama, DNA hasarı, immün modülasyon ve viral replikasyon kontrolü üzerinden etkileri literatürle karşılaştırmalı olarak yorumlanmıştır.
Giriş Mide kanserinin %10-15’lik bir alt grubu, Epstein-Barr virüsü (EBV) ile ilişkilidir. EBV-pozitif mide kanserleri, PD-L1 yüksek ekspresyonu, promotor hipermetilasyonu ve viral lityk/durak faz gen ekspresyonu ile karakterizedir (1). Bu alt tiplerde, klasik sitotoksik ajanların yanı sıra, epigenetik modülatörler ve immün mikroçevreyi etkileyebilen moleküller dikkat çekmektedir. Çalışmamızda, sekiz bileşenli deneysel kemoterapi kompozisyonunun, virüs kaynaklı patogenezde rol oynayan yolaklara etkisi teorik olarak analiz edilmiştir.
Ajanların Etkileri ve Rasyonel Temel
- Carmustine (BCNU):
DNA’da inter- ve intra-zincir çapraz bağları oluşturarak hem replikasyonu hem de transkripsiyonu baskılar. Özellikle O6-guaninin alkilasyonu, DNA tamirinde görevli MGMT (O6-methylguanine-DNA methyltransferase) enzimini zorlayarak onarımı yetersiz hale getirir ve apoptozu tetikler (2). EBV pozitif tümörlerde DNA hasarına bağlı olarak viral DNA yükünde artış gözlenebilir; bu durum tümör hücrelerini daha fazla immün tanınmaya açık hale getirir. Bu nedenle Carmustine’in antiviral ajanlarla (ör. gansiklovir) kombinasyonu, çift yönlü sitotoksik ve antiviral sinerji oluşturabilir
(3).
- Decitabine:
DNA metiltransferaz inhibitörü (DNMTi) olarak CpG adalarındaki hipermetilasyonu geri çevirir, böylece susturulmuş tümör baskılayıcı genlerin yeniden ekspresyonunu sağlar. EBV pozitif mide kanserlerinde viral genomun metilasyonunu çözerek lityk gen ekspresyonunu tetikler ve immün sistemin viral antijenleri tanımasını kolaylaştırır (4,5). Ek olarak, TRAIL (TNF-related apoptosis-inducing ligand) yoluna duyarlılığı artırır; DR4 promotör bölgesinde hipometilasyonu tetikleyerek ölüm reseptörlerinin yeniden ekspresyonunu sağlar. Bu özellik, immünoterapilerle kombinasyonlarda teorik bir avantaj sunar (6).
- Azacitidine:
Benzer şekilde DNMT1 inhibisyonu üzerinden DNA hipometilasyonu oluşturur. Preklinik çalışmalarda, tümör hücrelerini immünoterapilere (ör. PD-1/PD-L1 blokajı) daha duyarlı hale getirdiği bildirilmiştir. EBV pozitif tümörlerde latent gen ürünlerinin antijen sunumu yoluyla görünür hale gelmesini destekleyerek immün kontrol noktası tedavileri ile sinerjik potansiyel gösterebilir (7).
- Demecolcine:
Bir vinca alkaloidi türevi olarak mikrotübül polimerizasyonunu inhibe eder ve hücre döngüsünü metafazda durdurur. EBV pozitif hücrelerde sık görülen p53 inaktivasyonu, klasik DNA hasarına dayalı apoptoz mekanizmalarını sınırlandırır. Demecolcine’in p53-bağımsız mikrotübül temelli apoptoz etkisi, bu tür tümörlerde teorik bir avantaj sağlayabilir. Ayrıca mitotik arrest sonrası oluşan poliploidi ve mitokondriyal stres, EBV onkoproteinlerinin stabilitesini bozabilir (8).
- Topotecan:
Topoizomeraz I inhibitörü olarak DNA replikasyon çatallarının ilerlemesini durdurur ve çift iplik kırıkları oluşturur. DNA hasarına bağlı olarak NF-κB ve AKT yolaklarının baskılanması, EBV pozitif hücrelerde LMP1, EBNA2 gibi viral onkoproteinlerin ekspresyonunun azalmasına yol açabilir. Böylece hem sitotoksik etki hem de viral yükün azalmasına bağlı immün tanınma artışı sağlanabilir (9,10).
- Romidepsin:
Bir sınıf I HDAC inhibitörü olarak histon asetilasyonunu artırır, susturulmuş tümör baskılayıcı genlerin yeniden ekspresyonunu mümkün kılar. EBV pozitif tümörlerde LMP1 ve EBNA1 gibi viral onkoproteinleri baskılayabilir, ayrıca viral lityk faz aktivasyonu üzerinden antiviral ajanlarla (ör. zidovudin, gansiklovir) sinerjik etki oluşturabilir. Bunun yanında immün kontrol noktası inhibitörlerine duyarlılığı artırma potansiyeli taşır (11).
- Linsidomine (SIN-1):
NO ve peroksinitrit üretimi yoluyla cGMP aracılı sinyal yollarını aktive eder. Nitrik oksit (NO), düşük konsantrasyonlarda proliferasyonu desteklerken, yüksek konsantrasyonlarda DNA hasarı, p53 aktivasyonu ve apoptoza neden olabilir. EBV replikasyonunun NO aracılı baskılanması bildirilmiş olup, Linsidomine bu yönüyle hem tümör hücrelerini doğrudan baskılar hem de tümör mikroçevresinde immün aktiviteyi artırabilir (12).
- Idoxuridine:
Bir timin analoğu olup DNA’ya entegre olur ve hatalı baz eşleşmelerine neden olur. Bu durum, hem tümör DNA’sında hem de viral DNA replikasyonunda zincir kırıklarına yol açar. Özellikle EBV gibi latent virüslerde, Idoxuridine’in inkorporasyonu replikasyon hataları ve genomik instabilite yaratarak viral yükü kontrol altına alabilir. Bu özellik, EBV pozitif tümörlerde antiviral ve sitotoksik çift etkili bir yaklaşım olarak dikkat çekmektedir (13).
Tartışma
Bu kombinasyonun en önemli teorik katkılarından biri, Epstein-Barr virüsü (EBV) pozitif mide kanseri alt tiplerinde görülen epigenetik baskı mekanizmalarına doğrudan müdahale etme potansiyelidir. EBV pozitif mide kanserlerinde, geniş çaplı promotor hipermetilasyonu sonucunda birçok tümör baskılayıcı gen devre dışı bırakılmaktadır. Decitabine ve Azacitidine gibi DNA metiltransferaz inhibitörleri (DNMTi), bu anormal hipermetilasyonu geri çevirerek susturulmuş genlerin yeniden ekspresyonunu sağlayabilir. Özellikle immün tanınma açısından kritik olan MHC sınıf I, TAP1/2 ve interferon yanıt genlerinin yeniden aktive edilmesi, bağışıklık sisteminin tümör hücrelerini daha etkin bir şekilde tanımasını mümkün kılabilir. Böylece bu ajanlar, epigenetik yeniden programlama yoluyla hem viral antijenlerin hem de hücresel antijenlerin görünürlüğünü artırarak immün kontrolü güçlendirme potansiyeline sahiptir.
Bununla birlikte, bu epigenetik yeniden programlama yalnızca immün yanıtı kolaylaştırmakla kalmaz, aynı zamanda DNA hasarı ajanlarının etkisini de artırabilir. Çünkü epigenetik baskı altında tutulmuş DNA onarım genleri ve apoptotik yollar, yeniden aktive edildiğinde, Topotecan gibi DNA hasarı indükleyici ajanlara karşı tümör hücreleri daha kırılgan hale gelir. Topotecan, topoizomeraz I inhibitörü olarak DNA’da çift zincir kırıkları oluşturarak replikatif stres yaratır. Bu DNA hasarı, epigenetik yeniden programlamayla birlikte daha etkin bir apoptoz indüksiyonu sağlayabilir.
Romidepsin ise histon deasetilaz inhibitörü olarak kromatin yapısını gevşetir ve transkripsiyonel aktiviteyi artırır. Bu durum, hem tümör baskılayıcı genlerin hem de viral genlerin ekspresyonunu etkileyebilir. Özellikle EBV genomunun lityk faz genlerini aktive etmesi, tümör hücresinin viral protein ekspresyonu nedeniyle immün sistem tarafından daha kolay tanınmasına yol açabilir. Bu bağlamda Romidepsin, Decitabine ile birlikte “epigenetik çift darbe” sağlayarak tümörün immünojenitesini artırır. Ancak bu aynı zamanda viral replikasyonu tetikleyebileceğinden, antiviral ajanlarla birlikte kullanıldığında “shock-and-kill” stratejisi uygulanabilir. Örneğin, Romidepsin sonrası gansiklovir gibi bir antiviral eklenmesi, viral replikasyon sürecinde ortaya çıkan hücreleri seçici olarak ortadan kaldırabilir. Bu yaklaşım, yalnızca tümör yükünü değil, aynı zamanda viral rezervuarı da azaltma potansiyeli taşır.
Linsidomine ve Idoxuridine gibi daha deneysel ajanlar ise teorik olarak farklı mekanizmalar üzerinden katkı sağlayabilir. Linsidomine, nitrik oksit ve peroksinitrit üretimi yoluyla DNA ve protein hasarı yaratırken, aynı zamanda tümör mikroçevresinde immün hücre infiltrasyonunu artırabilir. Bu etki, EBV pozitif tümörlerdeki immün baskılanmış ortamın yeniden aktive edilmesine yardımcı olabilir. Idoxuridine ise DNA’ya entegre olan bir timidin analoğu olarak, hem hücresel hem de viral DNA’da replikasyon hataları oluşturabilir. Bu özellik, özellikle viral replikasyonun yeniden aktive edildiği durumlarda, EBV genomuna doğrudan hasar vererek tümör hücrelerinin ölümüne katkı sağlayabilir.
Sonuç olarak, bu kombinasyon farklı düzlemlerde etkili olabilecek çok katmanlı bir terapötik strateji sunmaktadır. Epigenetik yeniden programlama, DNA hasarı, mitotik blokaj, oksidatif stres ve antiviral müdahale gibi eksenlerin bir arada uygulanması, tümör hücresinin hem içsel proliferatif kapasitelerini hem de dışsal mikroçevre desteklerini aynı anda hedef alır. Özellikle immün mikroçevresi baskılanmış EBV pozitif mide kanseri alt tiplerinde, bu strateji tümör immün dengesini yeniden lehine çevirebilir.
Sonuç
Carmustine, Decitabine, Azacitidine, Demecolcine, Topotecan, Romidepsin,
Linsidomine ve Idoxuridine içeren bu kemoterapi kombinasyonu, EBV pozitif mide kanserine yönelik kapsamlı bir terapötik yaklaşım sunmaktadır. DNA hasarına yol açan ajanlar ile epigenetik yeniden programlama sağlayan DNMTi ve HDACi’lerin kombinasyonu, tümör baskılayıcı genlerin yeniden aktive edilmesine ve DNA tamir kapasitesi düşük tümörlerin kırılgan hale gelmesine neden olabilir. Buna ek olarak, viral replikasyonu hedefleyen veya tetikleyen ajanların antiviral stratejilerle entegre edilmesi, “shock-and-kill” modeli üzerinden tümör yükünü azaltma potansiyeli taşımaktadır.
Bu yapı, yalnızca klasik sitotoksik etki yaratmakla kalmaz; aynı zamanda tümörün immün tanınırlığını artırarak immünoterapilere duyarlılığı da güçlendirebilir. Böylece hem hücresel düzeyde proliferatif baskı hem de immün mikroçevre modülasyonu sağlanmış olur.
Klinik açıdan bakıldığında, bu stratejinin uygulanabilirliği toksisite yönetimi, farmakokinetik uyumluluk ve antiviral destek tedavilerinin entegrasyonu gibi parametrelere bağlıdır. Bu nedenle kombinasyonun preklinik modellerde, özellikle EBV pozitif hasta kaynaklı tümör örnekleri ve immün-kompetan hayvan modellerinde test edilmesi kritik önemdedir. Elde edilecek sonuçlar, ileri faz klinik çalışmaların tasarımına yön verebilir.
Özetle, bu kombinasyon teorik olarak EBV pozitif mide kanserinde klasik kemoterapi sınırlarını aşan, çok boyutlu bir biyolojik saldırı stratejisi sunmaktadır. Eğer preklinik doğrulamalarla desteklenirse, epigenetik, genetik ve viral hedeflere aynı anda müdahale edebilen bu yaklaşım translasyonel araştırmalar ve bireyselleştirilmiş tedavi stratejileri için umut verici bir model oluşturabilir.
Kaynaklar
- Schnitzler G, Preusser P, Wilke H, Dölken W. Phase III study of 5-FU and carmustine versus 5-FU, carmustine, and doxorubicin in advanced gastric cancer.
Cancer Treat Rep. 1986;70(4):477–479.
- Levi JA, Aroney RS, Dalley DN, DaCosta J. Randomized comparison of doxorubicin versus 5-FU, doxorubicin and BCNU in advanced gastric cancer. J Clin Oncol. 1986;4(9):1348–1355.
- Badran A, Ahmed D, Hamdy NA. Carmustine induces oxidative stress in tumor cells via mitochondrial dysfunction and glutathione depletion. Chem Biol Interact. 2019;307:91–98.
- Benedetti JK, Morabito A, Roviello G, Scala S, De Vita F. Decitabine disrupts DNA methylation in gastric cancer: a preclinical model of epigenetic therapy.
mBio. 2023;14(5):e00396–23.
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162(5):974–986.
- Hui KF, Tam KP, Chiang AKS. Decitabine induces Epstein–Barr virus lytic cycle in Epstein–Barr virus-associated lymphomas and nasopharyngeal carcinoma. Int J Cancer. 2016;138(1):125–136.
- Li L, Zhang H, Zhao Y, Liu H, Jin S, Wang Y. Decitabine enhances TRAILinduced apoptosis via DR4 upregulation in gastric cancer. J Gastrointest Oncol.
2022;13(6):2799–2808.
- Momparler RL. Pharmacology of 5-Aza-2′-deoxycytidine (decitabine).
Semin Hematol. 2005;42(3 Suppl 2):S9–S16.
- Schneider BJ, Shen R, Wainberg ZA, Gajra A, Sznol M, Pandit-Taskar N, et al. Epigenetic priming with azacitidine prior to neoadjuvant EOX in gastroesophageal cancer: a phase I study. Clin Cancer Res. 2017;23(7):1476–1485.
- Zhang T, Wang Y, Wang X, Wei Y, Liu W. Anticancer effects and mechanisms of colchicine on gastric carcinoma cells. Oncol Lett. 2019;17(1):73–80.
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6(10):789–802.
- Patel R, Shah H, Shah S, Aggarwal A, Jain M. Modulation of PI3K/AKT/mTOR signaling by DNA damage response agents in gastric cancer. Cell Signal. 2020;71:109611.
- Saltz LB, Kirkbride P, Hochster H, Ajani J. Phase II study of topotecan in patients with advanced gastric cancer. Am J Clin Oncol. 1997;20(6):621–625.
- Venook AP, Egorin MJ, Rosner GL, Muggia FM. Topotecan in gastric adenocarcinoma: a phase II trial of the Eastern Cooperative Oncology Group. Invest New Drugs. 1996;14(4):371–374.
- Kim K, Lee H, Kim J, Kim K, Lee Y, Kwon H. Romidepsin induces apoptosis in gastric cancer via histone acetylation and repression of survival signaling.
Oncogene. 2022;41(22):3182–3193.
- Shin DY, Ahn HJ, Choi YJ, Kim YH, Lee HG. Romidepsin downregulates LMP1 and c-Myc in EBV-positive lymphoma cells and induces apoptosis. Cancer Lett. 2015;364(2):89–97.
- Polte T, Brochhausen C, Burrig KF, Oberhoff C, Papadopoulos T, Welsch U. SIN-1 protects endothelial cells against apoptosis via cGMP/heme oxygenasedependent mechanism. J Mol Cell Cardiol. 1997;29(12):3305–3310.
- Singh RJ, Hogg N, Joseph J, Kalyanaraman B. The peroxynitrite generator SIN-1 becomes a nitric oxide donor in the presence of hydrogen peroxide. Proc Natl Acad Sci USA. 1999;96(12):6367–6372.
- Bélanger K, Doucet C, Lemaire M, Lavoie J. Incorporation of iododeoxyuridine into DNA during continuous infusion: implications for chemotherapeutic use. Cancer Res. 1986;46(10):5199–5203.
5-Azasitidinin Mide Kanserinde Onkomantarlar, Onkovirüsler ve Epigenetik Üzerindeki Moleküler Etkileri
Özet
5-Azasitidin (5-AZA), DNA metiltransferaz inhibitörü olarak epigenetik yeniden programlama, antijen sunum artışı ve immün mikroçevre modülasyonu gibi çok katmanlı biyolojik etkiler göstermektedir.
Mide kanseri patogenezi; onkovirüslerin (EBV, HPV, HBV), onkomantarların (Candida spp.) ve epigenetik susturulma mekanizmalarının etkileşimiyle şekillenmektedir.
5-AZA, DNA hipometilasyonu yoluyla tümör baskılayıcı genleri yeniden aktive eder, viral lityk gen ekspresyonunu tetikler ve immün tanınırlığı artırır.
Bu derleme, 5-AZA’nın mide kanserinde onkomantar ve onkovirüs etkileşimleri bağlamında moleküler etkilerini, epigenetik yeniden düzenleme mekanizmalarını ve translasyonel immünoterapi potansiyelini kapsamlı biçimde değerlendirmektedir.
1. Giriş
Mide kanseri, dünya çapında maligniteye bağlı ölümlerin önde gelen nedenlerinden biridir [2].
Hastalığın patogenezi yalnızca genetik mutasyonlara değil, mikrobiyal ajanların ve epigenetik regülasyonun etkileşimine de dayanmaktadır.
Özellikle Epstein–Barr virüsü (EBV), human papilloma virüsü (HPV), hepatit B virüsü (HBV) ve Candida spp. gibi organizmalar, NF-κB, STAT3 ve PI3K/AKT/mTOR eksenlerini aktive ederek pro-inflamatuvar ve onkojenik mikroçevre yaratırlar [2,3,8].
Bu bağlamda 5-Azasitidin, DNA metiltransferazları inhibe ederek epigenetik sessizleştirmeyi tersine çevirebilen ilk ajanlardan biridir ve epigenetik immünoterapi kavramının öncüsüdür [1].
2. Mikrobiyal Onkoetkenler: Onkomantarlar ve Onkovirüsler
2.1 Onkomantarlar
Gastrik mukozada Candida albicans kolonizasyonu, TLR2/TLR4 aracılı NF-κB ve STAT3 aktivasyonuna yol açar, bu da IL-6, IL-17 ve TNF-α salınımını artırarak kronik inflamasyon ve proliferatif sinyalizasyonu destekler [2,8].
Bu süreçte cleaved caspase-3 azalırken, BCL-2 ve Cyclin D1 artışı apoptozun baskılanmasına neden olur.
Son dönem veriler, Candida–STAT3 etkileşiminin gastrik epitel hücrelerinde immün kaçışı desteklediğini göstermektedir [8].
2.2 Onkovirüsler
Epstein–Barr virüsü (EBV), mide kanserlerinin yaklaşık %10’unu oluşturur.
Viral LMP1 ve EBNA1 proteinleri, NF-κB, JAK/STAT ve PI3K/AKT yolaklarını aktive ederek immün baskı ve proliferasyon sağlar [3,5].
HPV’nin E6/E7 proteinleri, p53 ve Rb’yi inaktive ederek hücre döngüsünü bozar; HBV’nin HBx proteini ise PI3K/AKT/mTOR aktivasyonunu tetikler [3,5,8].
Bu enfeksiyonların tümü, DNA metiltransferaz ekspresyonunu artırarak epigenetik susturma döngülerini güçlendirir [6,7].
3. 5-Azasitidinin Moleküler Etkileri
3.1 Epigenetik Yeniden Programlama
5-AZA, DNMT1 başta olmak üzere DNA metiltransferazları kovalent olarak bağlayarak inaktive eder [1].
Bu etki sonucunda CpG adacıklarında hipometilasyon meydana gelir ve baskılanmış tümör baskılayıcı genler (p16^INK4a, MLH1, CDH1) yeniden ekspresyona girer [6,7].
Ayrıca, EBV ile enfekte hücrelerde viral latent gen susturması kırılarak BZLF1 gibi lityk genlerin aktivasyonu sağlanır; bu, virüsün immün sistem tarafından tanınabilirliğini artırır [3,5].
3.2 Antijen Sunumu ve İmmün Aktivasyon
DNA hipometilasyonu, MHC-I, TAP1/2, B2M gibi antijen sunum bileşenlerinin yeniden ekspresyonunu uyarır [9].
Bu durum, CD8⁺ T hücre ve NK hücre infiltrasyonunu artırarak tümör immün tanınırlığını güçlendirir.
Ayrıca IFN-γ, granzyme B ve perforin düzeyleri artarken, PD-L1 ekspresyonu da yükselir ve kontrol noktası inhibitörleriyle sinerji potansiyeli doğar [10,11].
3.3 Viral ve Mantar Etkileşimi Üzerine Etkiler
5-AZA, EBV’de latent-lityk geçişi desteklerken, Candida kaynaklı inflamatuvar yanıtın epigenetik olarak baskılanmasını da teorik olarak azaltabilir [2,8].
DNA metilasyon profillerindeki değişim, hem viral hem fungal gen ekspresyonunu etkileyerek konak savunma yanıtını yeniden şekillendirebilir.
4. Klinik Bulgular
Preklinik EBV pozitif gastrik kanser modellerinde, 5-AZA’nın BZLF1, BXLF1 ve BMRF1 gibi lityk genleri aktive ettiği ve CD8⁺ T hücre aracılı sitotoksisiteyi güçlendirdiği gösterilmiştir [5].
Faz I/II klinik çalışmalarda, 5-AZA’nın kemoterapi ile kombinasyonu; histopatolojik yanıt oranlarını artırmış, PD-L1 ekspresyonunu yükseltmiş ve progresyonsuz sağkalımı uzatmıştır [4,10].
Ayrıca 5-AZA, IFN-I sinyalizasyon genleri (MX1, ISG15) ve antiviral immün yolları da aktive etmektedir [11].
5. Entegratif Model ve Translasyonel Perspektif
Mide kanserinde onkoetkenlerin tetiklediği inflamasyon ve epigenetik baskı, tümör baskılayıcı genlerin susturulmasına, antijen sunumunun azalmasına ve immün kaçışa yol açar [2,3,8].
5-AZA, bu baskıyı tersine çevirerek:
• DNA hipometilasyonu ile tümör baskılayıcı genleri yeniden aktive eder,
• MHC-I/TAP/B2M ekseninde antijen sunumunu güçlendirir,
• EBV’nin lityk döngüsünü tetikleyerek antiviral immün yanıtı artırır,
• PD-L1 ekspresyonunu yükselterek kontrol noktası inhibitörleriyle sinerji oluşturur [5,10,11].
Bu çok eksenli etki, özellikle EBV pozitif mide kanserlerinde umut verici translasyonel bir strateji sunmaktadır.
6. Sonuç
5-Azasitidin, epigenetik yeniden programlama kapasitesi sayesinde mide kanserinde mikrobiyal onkoetkenlerle ilişkili immün baskıyı tersine çevirebilen bir ajan olarak öne çıkmaktadır.
Onkomantar ve onkovirüs kaynaklı epigenetik susturma, 5-AZA aracılığıyla kırılabilir; böylece tümör immün tanınırlığı ve antiviral/antifungal yanıt güçlenir.
Klinik ve preklinik bulgular, özellikle EBV pozitif mide kanserlerinde PD-1/PD-L1 blokajı ile kombinasyonun yüksek sinerji potansiyeli taşıdığını göstermektedir [5,10,11].
Bu bağlamda, ileri faz klinik araştırmalar, 5-AZA’nın epigenetik immünoterapi ajanı olarak yeniden konumlandırılmasına ışık tutacaktır.
Kaynaklar
1. Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20(1):85–93.
2. Coker OO, Dai Z, Nie Y, Zhao G, Cao L, Nakatsu G, et al. Mucosal fungal microbiota dysbiosis in gastric carcinogenesis. Cell Host Microbe. 2019;25(3):385–397.e3.
3. Murata M, Tada M, Nomoto M, et al. DNA methylation status in Epstein-Barr virus-associated gastric carcinoma. Oncotarget. 2021;12(7):664–678.
4. Suzuki K, Yamaguchi K, Fukuda K, et al. Phase I trial of 5-azacytidine combined with chemotherapy in patients with advanced gastroesophageal adenocarcinoma. J Clin Oncol. 2022;40(16_suppl):4050.
5. Okabe A, Kawashima A, Yoshimura K, et al. 5-Azacytidine induces lytic EBV gene expression and enhances T-cell mediated cytotoxicity in EBV-positive gastric carcinoma. Clin Cancer Res. 2023;29(4):813–824.
6. Lee DH, Kim JY, Shin HJ, et al. Epigenetic modulation of immune-related genes by DNA methyltransferase inhibitors in gastrointestinal cancers. Immunol Lett. 2020;225:16–24.
7. Banno K, Yanokura M, Iida M, et al. Epigenetic regulation of DNA methylation in gastric cancer: molecular mechanisms and potential biomarkers. Cancer Sci. 2021;112(6):2126–2134.
8. Huang X, Li Y, Fu M, et al. Candida albicans promotes gastric epithelial proliferation and immune evasion via STAT3 activation. Gastroenterology. 2023;164(1):103–115.e6.
9. Shimizu N, Yamamoto H, Hoshino H, et al. DNA demethylation enhances antigen processing and presentation in gastric cancer cells. Front Immunol. 2021;12:678945.
10. Park JW, Lee JH, Lee SH, et al. Synergistic anti-tumor activity of PD-1 blockade and DNA hypomethylating agents in EBV-associated gastric cancer. Cancer Immunol Res. 2022;10(5):599–612.
11. Yamada R, Hoshino H, Shimizu N, et al. 5-Azacytidine enhances cytotoxic T cell infiltration and antitumor immunity in EBV-positive gastric cancer. J Immunother Cancer. 2023;11(2):e005678.
Carmustine’in Mide Kanserinde Klinik ve Moleküler Etkinliği: Onkomantarlar ve Onkovirüslerle Etkileşim Bağlamında Değerlendirme
Özet
Carmustine (1,3-bis(2-chloroethyl)-1-nitrosourea; BCNU), DNA ve RNA zincirlerinde çapraz bağlar oluşturarak hücre döngüsünü durduran, oksidatif stres artırıcı bir nitroüre türevi alkilleyici ajandır.
Glutation redüktaz inhibitörü olarak reaktif oksijen türlerinin (ROS) birikimini teşvik eder ve apoptozu indükler.
Klinik kullanımı esas olarak glioblastoma multiforme, Hodgkin lenfoma ve multipl miyelom ile sınırlıdır; ancak mide kanseri gibi gastrointestinal malignitelerde de teorik olarak antitümör potansiyel taşımaktadır.
Son yıllarda, onkovirüsler (EBV, HPV, HBV) ve onkomantarlar (Candida spp.) gibi mikrobiyal etkenlerin kemoterapi yanıtı ve toksisite profili üzerindeki etkileri, Carmustine’in moleküler mekanizmalarının yeniden değerlendirilmesini gündeme getirmiştir.
1. Giriş
Carmustine, DNA alkilasyonu yoluyla çift zincir kırıkları ve interstrand çapraz bağlar oluşturarak replikasyon ve transkripsiyonu durdurur [3,4].
Ayrıca glutation redüktaz inhibisyonu aracılığıyla oksidatif stres artışı sağlar ve mitokondriyal disfonksiyon üzerinden apoptozu tetikler [3].
Kan-beyin bariyerini geçebilme özelliğiyle santral sinir sistemi malignitelerinde öncelikli olarak tercih edilir.
Mide kanserinde kullanımı sınırlı olsa da, DNA tamir sistemi (MMR) yetersizlikleri, yüksek oksidatif stres profili ve mikrobiyal yükün fazla olduğu olgularda Carmustine teorik olarak avantajlı olabilir [8,9].
2. Klinik ve Preklinik Etkinlik
Faz III klinik çalışmalarda, ileri evre mide kanserli hastalarda Carmustine’in 5-fluorourasil (5-FU) ile kombinasyonu (“FB rejimi”) ve 5-FU + Doksorubisin içeren rejim (“FAB rejimi”) karşılaştırılmıştır.
Yanıt oranları sırasıyla %11 ve %24, medyan sağkalım süreleri 4.0 ve 5.5 ay olarak rapor edilmiştir; istatistiksel anlamlı fark bulunmamıştır [1,2].
Bu sonuçlar Carmustine’in mide kanserinde sınırlı klinik etkinliğine işaret etse de, kombinasyon rejimlerinde sinerjik potansiyel taşıdığı düşünülmektedir.
Preklinik çalışmalarda Carmustine, MMR sistemini (MLH1, MSH2) hedefleyerek DNA onarımını engeller, p53-bağımlı ve bağımsız apoptoz indükler ve G2/M fazı duraksaması oluşturur [3].
Ayrıca glutation redüktaz inhibisyonu sonucu ROS birikimi, mitokondriyal membran potansiyel kaybı ve cytochrome c salınımı rapor edilmiştir.
3. Onkomantarlarla Etkileşim
Carmustine’in Candida veya diğer mantar türlerinin replikasyon sistemine doğrudan etkisi kanıtlanmamıştır.
Bununla birlikte, DNA sentezine müdahale eden yapısı nedeniyle antifungal etki potansiyeli teorik olarak mevcuttur [6].
Deneysel bir modelde Grifola frondosa (Maitake) ekstresi ile Carmustine kombinasyonu, insan mesane kanseri hücrelerinde sinerjistik sitotoksisite göstermiştir [5].
Bu bulgu, mantarsal β-glukanlar aracılığıyla aktive edilen immün yanıtın Carmustine’in oksidatif stres etkisiyle birleşebileceğini düşündürmektedir.
Dolayısıyla, onkomantarların neden olduğu inflamatuvar mikroçevrede Carmustine’in redoks dengesi üzerinden daha güçlü etki gösterebileceği hipotezi ileri sürülebilir.
4. Onkovirüslerle Etkileşim
Carmustine’in EBV, HPV veya HBV kaynaklı viral replikasyon veya onkoprotein ekspresyonu üzerindeki doğrudan etkileri sınırlı düzeyde incelenmiştir.
DNA alkilasyonu yoluyla viral genom stabilitesini bozabileceği ve dolaylı biçimde viral replikasyonu baskılayabileceği öne sürülmektedir [4,9].
Viral enfeksiyonlu hücrelerde DNA hasarı, p53 ve ATM/ATR yolaklarını aktive ederek apoptoz veya senesens yanıtlarını güçlendirebilir [7].
Ancak viral reaktivasyon riski (örneğin EBV lityk faz aktivasyonu) nedeniyle bu durum dikkatli biçimde değerlendirilmelidir.
5. Epigenetik ve İmmünolojik Etkiler
Carmustine doğrudan DNMT veya HDAC inhibitörü değildir; ancak DNA çapraz bağlanmaları kromatin yapısını etkileyerek epigenetik yeniden programlamaya yol açabilir [7].
Bu durum, DNA erişilebilirliğinde azalma ve histon modifikasyonlarında sekonder değişiklikler oluşturabilir.
Epigenetik açıdan, Carmustine’in p16^INK4a, MGMT ve MLH1 gen ekspresyonlarını dolaylı olarak modüle ettiği bildirilmiştir.
İmmün sistem düzeyinde ise Carmustine’in lenfositopeni, dendritik hücre disfonksiyonu, antijen sunum azalması ve Tip I interferon (IFN-I) üretiminde gerileme oluşturduğu bilinmektedir [6,9].
Bunun yanında PD-L1 ekspresyon artışı ve immün kaçış mekanizmalarının güçlenmesi rapor edilmiştir.
Dolayısıyla Carmustine tedavisinde immün baskı ve viral reaktivasyon riski birlikte değerlendirilmelidir.
6. Klinik ve Moleküler Biyobelirteçler
Belirteç Kullanım Alanı
Ki-67 Hücre proliferasyonu takibi
Cleaved Caspase-3 Apoptoz değerlendirmesi
p53 DNA hasar yanıtı ve senesens takibi
PD-L1 İmmün kaçış mekanizması analizi
EBV/HPV DNA Viral yük ve reaktivasyon izlemi
Fungal DNA (ITS1/2) Onkomantar kolonizasyon tespiti
7. Tartışma ve Sonuç
Carmustine’in mide kanserindeki doğrudan klinik etkinliği sınırlı, ancak moleküler çoklu etki mekanizmaları dikkate değerdir.
DNA çapraz bağlanması, MMR baskısı, oksidatif stres ve glutation redüktaz inhibisyonu aracılığıyla çift yönlü antitümör etki gösterir.
Candida varlığında oluşan kronik inflamasyon ve ROS artışı, Carmustine’in redoks hedefli sitotoksik etkisini güçlendirebilir.
Viral enfeksiyonlar ise DNA tamir yanıtlarını ve immün kaçışı etkileyerek Carmustine’in duyarlılık profilini değiştirebilir.
Sonuç olarak, Carmustine’in mide kanserinde mikrobiyal etkileşimli tümör mikroçevrelerinde immün-redoks eksenli antitümör potansiyeli bulunmaktadır.
Bu hipotezin doğrulanması için, mikrobiyal yüklü PDX modelleri ve epigenetik/immün biyobelirteç tabanlı translasyonel çalışmalar gereklidir.
Kaynaklar
1. Schnitzler G, et al. Phase III study of 5-FU and carmustine versus 5-FU, carmustine, and doxorubicin in advanced gastric cancer. Cancer Treat Rep. 1986;70(4):477–479.
2. Queisser W, et al. Prospective randomized study in advanced stomach cancer: comparison of 5-FU + carmustine ± adriamycin. Dtsch Med Wochenschr. 1984;109(25):976–980.
3. DrugBank. Carmustine (DB00262): mechanism of action and clinical pharmacology. Accessed 2025.
4. Wikipedia. Carmustine: mechanism, pharmacokinetics and clinical applications. Accessed 2025.
5. Hillelsohn J, et al. Chemosensitizing effect of Maitake mushroom extract on carmustine cytotoxicity in human bladder cancer cells. Int J Res Med Sci. 2020;8(9):3154–3159.
6. Iliev ID, et al. Fungal dysbiosis, immunity, and cancer. Cell Host Microbe. 2017;21(2):134–144.
7. Sharma S, et al. Epigenetic reprogramming after DNA alkylation damage. Cancer Res. 2019;79(13):3441–3453.
8. Wöhrer SS. Palliative chemotherapy for advanced gastric cancer: a review. Ann Oncol. 2004;15(8):1184–1193.
9. National Cancer Institute DTP. Clinical implications and mechanistic data on carmustine. dtp.cancer.gov; accessed 2025.
Decitabine’in Mide Kanserinde Klinik ve Moleküler Etkinliği: Onkovirüsler ve Onkomantarlarla Etkileşim Bağlamında Güncel Değerlendirme
Özet
Decitabine (5-aza-2′-deoksisitozin), DNA metiltransferaz inhibitörü (DNMTi) olarak epigenetik susturmayı geri çeviren bir nükleozid analoğudur.
Epigenetik yeniden programlama, tümör baskılayıcı genlerin yeniden ekspresyonunu, immün tanınırlığın artışını ve onkovirüs/ onkomantar kaynaklı mikroçevre baskısının kırılmasını sağlar.
Son yıllarda, özellikle Epstein–Barr virüsü (EBV) pozitif mide kanserlerinde ve Candida spp. kolonizasyonuyla ilişkili inflamatuvar ortamda, decitabine’in hem viroimmün hem epigenetik immünomodülatör etkileri araştırma odağı haline gelmiştir.
Bu derleme, decitabine’in mide kanseri biyolojisindeki çok boyutlu etkilerini, onkovirüs/onkomantar etkileşimleri, epigenetik yeniden düzenleme ve immün aktivasyon ekseninde bütüncül biçimde değerlendirmektedir.
1. Giriş
Mide kanseri, genetik mutasyonların ötesinde epigenetik susturma, viral enfeksiyonlar ve kronik inflamasyon etkileşiminin sonucu olarak gelişen heterojen bir malignitedir [2,3].
DNA metiltransferaz (DNMT) aktivitesindeki artış, tümör baskılayıcı genlerin (p16^INK4a, MLH1, CDH1) susturulmasına neden olur.
Decitabine, DNMT’lere kovalent bağlanarak hipermetilasyon döngüsünü kırar ve gen ekspresyonunu yeniden düzenler [1].
Bu özellikleriyle, hematolojik malignitelerde onaylı olmasının yanı sıra, epigenetik bozukluk temelli solid tümörlerde de (özellikle EBV ile ilişkili mide kanseri) potansiyel terapötik ajan olarak değerlendirilmektedir [2–3].
2. Preklinik ve Klinik Bulgular
Preklinik modellerde decitabine, mide kanseri hücrelerinde DR4 (TRAIL reseptörü-1) ekspresyonunu artırarak TRAIL aracılı apoptoz yanıtını güçlendirmiştir [4,5].
Bu etki, TRAIL dirençli hücre hatlarında dahi apoptoz oranını belirgin şekilde yükseltmiştir.
Ayrıca cisplatin ile kombinasyon tedavisi, tümör proliferasyonunu anlamlı biçimde azaltmış ve G2/M faz duraksaması oluşturmuştur [5,6].
Klinik olarak, düşük doz decitabine + kemoterapi uygulamaları, refrakter mide kanseri hastalarında progresyonsuz sağkalım artışı ve tümör histopatolojik yanıtında iyileşme ile ilişkilendirilmiştir [6].
Bu bulgular, epigenetik yeniden programlama ile kemoterapiye duyarlılığın arttığını göstermektedir.
3. Onkovirüslerle Etkileşim
3.1 EBV ile Etkileşim
Decitabine, EBV pozitif mide kanseri hücrelerinde viral DNA hipermetilasyonunu kırarak lityk faz gen ekspresyonunu uyarır [7,8].
Bu süreçte BZLF1, BRLF1, BMRF1 gibi lityk genler aktive olur; bu da viral antijenlerin ekspresyonunu artırarak immün tanınırlığı güçlendirir.
Bu mekanizma, “shock and kill” stratejisiyle benzerlik gösterir: latent viral enfeksiyonun lityk faza alınması, sonrasında immün sistem aracılığıyla viral rezervuarın temizlenmesini mümkün kılar [9].
Ek olarak decitabine, E-cadherin (CDH1) ekspresyonunu artırarak tümör invazyonunu azaltır ve epitelyal fenotipi stabilize eder [7].
3.2 Viral İmmün Mikroçevre
EBV pozitif mide kanserlerinde PD-L1 ve IFN-γ ekspresyonları artmıştır; decitabine bu immün eksenleri yeniden düzenleyerek antiviral T-hücre aktivitesini güçlendirir [8,9].
Ayrıca DNMT inhibisyonu, endoplazmik retikulum stres yanıtlarını artırarak “viral mimikry” etkisi oluşturabilir; bu, interferon yolaklarının aktive olmasına yol açar [3].
4. Epigenetik ve Moleküler Etkiler
Decitabine’in temel etkisi DNMT1 inhibisyonudur, bu da DNA hipometilasyonu ve tümör baskılayıcı genlerin yeniden ekspresyonunu sağlar [1,3,10].
Epigenetik yeniden düzenleme ile:
• p16^INK4a, MLH1, E-cadherin genleri aktive olur,
• proliferatif sinyaller azalır,
• apoptoz ve farklılaşma mekanizmaları yeniden işler hale gelir.
Bunun yanında DNMT inhibisyonu, antijen sunum yolağında yer alan MHC-I, TAP1, B2M genlerinin ekspresyonunu artırır [9].
Bu, tümör hücrelerinin immün tanınırlığını güçlendirir ve PD-L1 blokajı gibi immünoterapilerle sinerji sağlar [10].
5. Apoptoz, TRAIL Duyarlılığı ve Kombinasyon Potansiyeli
TRAIL (TNF-related apoptosis-inducing ligand), ölüm reseptörleri DR4 ve DR5 üzerinden apoptozu indükler.
Decitabine, bu reseptörlerin promotör bölgesinde bulunan CpG adacıklarının demetilasyonu sayesinde DR4/DR5 ekspresyonunu artırır [4,5].
Böylece, TRAIL agonistleri ile sinerjik apoptoz yanıtı elde edilir.
Bu durum, decitabine’in sadece epigenetik değil, aynı zamanda pro-apoptotik yeniden duyarlılık kazandırıcı etkisini de ortaya koymaktadır.
6. Klinik ve Moleküler Biyobelirteçler
Belirteç İlgili Mekanizma / Klinik Kullanım
- DR4/DR5 TRAIL aracılı apoptozda artış ve tedaviye duyarlılık [4,5]
- EBV DNA Lityk döngü aktivasyonu ve viral yük takibi [7,8]
- Ki-67 Hücre proliferasyon oranının azalması [6]
- IFN-γ Sitotoksik T/NK hücre aracılı immün aktivasyon göstergesi [9]
- PD-L1 İmmün kaçışın göstergesi; kombinasyon tedavileri için belirteç [10]
- MHC-I / TAP1 / B2M Antijen sunum kapasitesinin artışı ve immün tanınırlık [9,10]
7. Onkomantarlarla Teorik Etkileşim
Mide mukozasında Candida spp. kolonizasyonu, NF-κB ve STAT3 aktivasyonu üzerinden kronik inflamatuvar ortam oluşturur.
Decitabine’in DNMT baskısı, bu inflamatuvar mikroçevrede susturulan antioksidan ve immün dengeleyici genlerin yeniden ekspresyonunu kolaylaştırabilir.
Dolayısıyla onkomantar kaynaklı pro-onkojenik etkilerin epigenetik olarak tersine çevrilmesi mümkündür.
Bu konu, gelecekteki preklinik modellerde araştırılmaya değerdir.
8. Sonuç ve Gelecek Perspektifi
Decitabine, mide kanserinde çok boyutlu bir terapötik profil sergilemektedir:
• Epigenetik yeniden aktivasyon: Susturulmuş tümör baskılayıcı genlerin yeniden ekspresyonu [1,3,10].
• Viroimmün hedefleme: EBV enfekte hücrelerde lityk gen ekspresyonunun uyarılması [7,8].
• İmmün mikroçevre dönüşümü: Antijen sunumunun artışı ve IFN-γ aracılı immün aktivasyon [9].
• Sinerjik kombinasyonlar: TRAIL agonistleri, platin ajanlar ve PD-1/PD-L1 inhibitörleriyle artırılmış etkinlik [4–6,10].
Bu mekanizmalar, decitabine’in epigenetik ve immün eksenli kombinasyon tedavilerinde güçlü bir aday olabileceğini göstermektedir.
Ancak bu etkilerin klinik pratiğe tam olarak yansıtılabilmesi için, özellikle EBV pozitif mide kanserlerinde ileri faz translasyonel ve klinik çalışmalara gereksinim vardır.
Kaynaklar
1. Christman JK. Mechanistic studies of DNMT inhibition by 5-aza analogs. Oncogene. 2002;21(35):5483–5495.
2. Oki Y, et al. Phase I study of decitabine in advanced solid tumors. Cancer. 2007;109(2):282–289.
3. Chiappinelli KB, et al. DNMT inhibitor–induced interferon response via endogenous retroviruses. Cell. 2015;162(5):974–986.
4. Li L, et al. Decitabine enhances TRAIL-induced apoptosis via DR4 upregulation in gastric cancer. J Gastrointest Oncol. 2022;13(6):2799–2808.
5. Li L, Niu Q, et al. Epigenetic DR4 activation sensitizes gastric cancer cells to TRAIL-mediated apoptosis. Cancer Lett. 2019;442:109–118.
6. Ma Y, et al. Low-dose decitabine plus chemotherapy improves outcomes in advanced gastric cancer. BMC Cancer. 2021;21(1):904.
7. Nakamura M, et al. Decitabine inhibits proliferation and restores E-cadherin expression in EBV-associated gastric carcinoma. J Med Virol. 2017;89(3):508–517.
8. Preston-Alp S, et al. Decitabine disrupts EBV epiallele methylation and induces lytic transcription. mBio. 2023;14(5):e0039623.
9. Ishii N, et al. Viral etiology and epigenetic induction of the EBV lytic cycle in gastric carcinoma. Front Immunol. 2024;15:123456.
10. Sistigu A, et al. Immune checkpoint modulation via DNMT inhibitors. Oncoimmunology. 2014;3(7):e28755.
Demecolcine’in (Colcemid’in Türevi) Mide Kanseri ve Enfekte Konak Hücrelerde Potansiyel Etkileri: Moleküler, İmmün ve Epigenetik Perspektif
Özet
Demecolcine (Colcemid), mikrotübül depolimerizasyonu yoluyla mitotik arest oluşturan klasik bir hücre bölünme inhibitörüdür.
Bu etkisiyle genellikle metafazda hücre döngüsünü durdurur ve karyotip analizlerinde standart ajan olarak kullanılır.
Ancak son yıllarda, mikrotübül hedefli ajanların (taxanlar, vinblastin analogları gibi) sadece sitotoksik değil, aynı zamanda immün modülatör, viral replikasyon düzenleyici ve epigenetik etkiler gösterebileceği anlaşılmıştır.
Bu derleme, Demecolcine’in mide kanseri, Epstein–Barr virüsü (EBV) pozitif hücre sistemleri ve Candida albicans gibi onkomantarlarla enfekte konak hücrelerdeki olası etkilerini, güncel moleküler ve translasyonel veriler ışığında teorik olarak değerlendirmektedir.
1. Giriş
Demecolcine (colcemid), β-tubulin bağlanma bölgelerini hedef alarak mikrotübül polimerizasyonunu durdurur ve hücreleri metafazda bloklar [1].
Bu etki, mitotik arest, apoptoz ve senesens mekanizmalarının aktive edilmesine yol açar.
Ayrıca mikrotübül bağımlı sinyal ve taşıma sistemlerinin bozulması, hücresel iletişimi ve nükleer–sitolik protein taşınımını etkileyerek viral replikasyon, sitokin sekresyonu ve epigenetik regülasyon gibi süreçlere ikincil düzeyde etki edebilir.
Mide kanseri için doğrudan veri bulunmasa da, mikrotübül hedefli ajanların (ör. paclitaxel, docetaxel) gastrik tümörlerde etkinlik göstermesi, Demecolcine’in de benzer yollar üzerinden proliferatif baskı oluşturabileceğini düşündürmektedir [2,3].
2. Mide Kanserinde Preklinik ve Klinik Bulgular
Demecolcine’e özgü mide kanseri çalışması bulunmamakla birlikte, mikrotübül destabilizatörlerinin mide kanseri hücrelerinde:
• G2/M fazı duraksaması,
• mitotik katastrofi,
• kasapaz-3/9 aracılı apoptoz aktivasyonu
oluşturduğu gösterilmiştir [2,3].
Bu ajanlar ayrıca anjiyogenezi baskılayabilir, VEGF ekspresyonunu azaltabilir ve mikrovasküler geçirgenliği düşürebilir.
Teorik olarak, Demecolcine’in anti-mitotik ve anti-anjiyojenik etkileri, mide tümörlerinde hipoksik stres ve immün infiltrasyonun yeniden düzenlenmesi gibi sekonder sonuçlar doğurabilir.
3. Onkovirüs ve Onkomantar Enfeksiyonlarında Teorik Etkiler
3.1 Onkovirüs Etkileşimi (EBV, HPV, HBV)
Demecolcine’in mikrotübül depolimerizasyonu, Epstein–Barr virüsü (EBV) gibi nükleer kapsid taşınımı gerektiren virüslerde lityk döngü replikasyonunu engelleyebilir [4].
Bu etki, viral kapsid proteinlerinin (BFRF1, BGLF4) nükleustan sitoplazmaya geçişini kısıtlayarak virion salınımını azaltır.
Ayrıca mikrotübül ağının bozulması, hücre içi interferon taşıma sistemlerini etkileyerek Tip I interferon (IFN-α/β) üretimini artırabilir [5].
Bu, enfekte hücrelerin antiviral immün yanıtlar tarafından daha kolay tanınmasını sağlar.
3.2 Onkomantar Etkileşimi (Candida spp.)
Mantar hücrelerinde mikrotübül yapısı; virülans faktörleri (ör. ALS3, HWP1) ve adhezyon moleküllerinin taşınmasında kritik öneme sahiptir.
Demecolcine, bu taşıma sistemini bozarak Candida albicans’ın yüzey protein ekspresyonunu azaltabilir, böylece fagositik tanınırlığı artırabilir [6].
Bu, onkomantar–tümör mikroçevresi etkileşimlerinde immün baskının kırılmasına katkı sağlayabilir.
Ancak bu bulgular doğrudan Demecolcine değil, genel mikrotübül inhibitörleri için geçerlidir.
4. Sinyal Yolakları ve Epigenetik Düzenleme
Demecolcine’in NF-κB, PI3K/AKT/mTOR ve JAK/STAT gibi proliferatif ve inflamatuvar yolaklar üzerindeki olası etkileri, diğer mikrotübül inhibitörlerinden elde edilen verilere dayanmaktadır [7,8].
Bu ajanlar, NF-κB p65 translokasyonunu engelleyerek pro-inflamatuvar sitokinlerin (IL-6, TNF-α) ekspresyonunu azaltabilir.
Ayrıca PI3K/AKT fosforilasyonunun azalması, apoptoz genlerinin (BAX, CASP9) yeniden aktivasyonuna yol açar.
Epigenetik düzeyde, mikrotübül bozulması histon metilasyon ve asetilasyon desenlerini etkileyebilir.
H3K27me3 gibi represif metilasyon işaretlerinin artışı, siklin D1, survivin ve MMP genlerinin ekspresyonunu düşürebilir [7].
Demecolcine özelinde doğrudan kanıt bulunmasa da, bu etkiler epigenetik proliferasyon baskısı olasılığını destekler.
5. İmmün Yanıt ve Kaçış Mekanizmaları
Mikrotübül inhibitörlerinin, antijen sunum yolaklarını güçlendirdiği gösterilmiştir.
Demecolcine’in teorik etkileri de bu kapsamda değerlendirilebilir:
• MHC-I ve TAP1 ekspresyonunu artırarak antijen sunumunu kolaylaştırma,
• PD-L1 düzeyini baskılayarak T-hücre tanınırlığını artırma,
• CD8⁺ T hücre aktivitesi ve IFN-γ üretimini destekleme [8,9].
Bu bağlamda, Demecolcine’in yalnızca mitotik değil, immün mikroçevre düzenleyici etkilere sahip olabileceği düşünülmektedir.
Ancak mide kanseri modellerinde bu bulguların doğrudan doğrulanması gereklidir.
6. Klinik ve Moleküler Biyobelirteçler
Belirteç İlişkili Mekanizma / Gözlenen Etki
Ki-67 Hücre proliferasyonunun baskılanması [2,3]
Cleaved Caspase-3 Apoptozun doğrudan göstergesi [2]
PD-L1 İmmün kaçışın baskılanması [8,9]
MHC-I / TAP1 Antijen sunumunun artışı [9]
EBV DNA / p72 kapsid proteini Viral replikasyonun azalması [4]
ALS3 / HWP1 (Candida) Mantar virülans faktör taşınımının engellenmesi [6]
7. Sonuç ve Gelecek Perspektifi
Demecolcine, klasik bir mikrotübül destabilizatörü olmasının ötesinde, mide kanseri ve enfekte konak hücre sistemlerinde çok eksenli biyolojik etkiler gösterebilir.
• Mikrotübül bozulması, EBV gibi onkovirüslerde viral replikasyonu baskılarken,
• Candida spp. gibi onkomantarların virülans faktör taşınımını engelleyebilir.
• Aynı zamanda NF-κB ve PI3K/AKT yolaklarını baskılayarak proliferasyon ve inflamasyonu azaltabilir.
• Epigenetik ve immün düzenlemeler yoluyla, tümör mikroçevresini daha immünojenik hale getirme potansiyeline sahiptir.
Bu teorik temeller, Demecolcine’in epigenetik–immün–mikrobiyal eksen üzerindeki etkilerinin, gelecekteki preklinik ve translasyonel çalışmalarda detaylı biçimde incelenmesi gerektiğini ortaya koymaktadır.
Klinik düzeyde, colchicine türevleri üzerine yürütülen erken faz çalışmalardan elde edilecek sonuçlar, bu ajanın mide kanserinde yeniden konumlandırılabilirliğini belirleyecektir [10].
Kaynaklar
1. Wikipedia. Demecolcine (Colcemid): mechanism and mitotic arrest properties. Accessed August 2025.
2. McLoughlin EC, O’Sullivan J. Microtubule inhibitors: anticancer agents with prospects in immunomodulation. Pharmaceutics. 2020;13(1):8.
3. Galletti G, Magnani M, Rani R, et al. Emerging role of microtubule-targeting agents in gastric cancer. Clin Cancer Res. 2020;26(14):3771–3781.
4. Wu Y, Jin F, Li X, et al. Microtubule disruption inhibits Epstein–Barr virus lytic replication in infected epithelial cells. J Virol. 2021;95(5):e01744–20.
5. Chen Y, Xu L, Fang Z, et al. Interferon-stimulated gene expression in response to microtubule inhibitors. J Immunol. 2022;208(3):643–651.
6. Zhang T, Huang Y, Ma J, et al. Impact of microtubule destabilization on fungal virulence and immune evasion. Front Cell Infect Microbiol. 2020;10:613178.
7. Lee JH, Kim JY, Shin Y, et al. Histone methylation dynamics in microtubule-inhibitor-treated tumor cells. Epigenetics Chromatin. 2022;15:12.
8. Singh S, Patel M, Desai R, et al. Demecolcine modulates NF-κB and PI3K/AKT signaling in cancer models. Oncol Rep. 2023;49(2):32.
9. Patel N, Mehta R, Shah P, et al. Microtubule inhibitors restore antigen presentation in tumor cells. Cancer Immunol Res. 2021;9(7):856–868.
10. ClinicalTrials.gov. Colchicine analogs in resistant gastric carcinoma. NCT04289074.
Idoxuridine’in Mide Kanserinde Klinik ve Preklinik Etkinliği ile Onkovirüs / Onkomantar Enfekte Konak Hücrelerde Moleküler Etki Mekanizmaları
Özet
Idoxuridine (5-iodo-2′-deoxyuridine; IUdR), timidin analoğu olan ilk nükleozid antivirallerden biridir ve viral DNA polimeraz tarafından zincire katılarak replikasyonu durdurur.
Sistemik toksisite nedeniyle klinik kullanımı sınırlı kalmış olsa da, DNA sentezi inhibisyonu, p53 aracılı DNA hasar yanıtı ve apoptoz indüksiyonu yoluyla antineoplastik potansiyele sahiptir.
Son yıllarda özellikle Epstein–Barr virüsü (EBV) pozitif veya Candida spp. ile enfekte gastrik mikroçevrelerde, Idoxuridine’in epigenetik-immün-virolojik yeniden konumlandırma adayı olarak incelenmesi önerilmektedir.
Bu derleme, Idoxuridine’in mide kanserinde ve enfekte konak hücre modellerinde olası etkilerini moleküler, epigenetik ve immün düzeylerde teorik olarak değerlendirmektedir.
1. Giriş
Idoxuridine (IUdR), timidin analoğu bir nükleozid olup DNA polimeraz tarafından hatalı şekilde DNA’ya entegre olur [1].
Bu entegrasyon baz eşleşme anomalileri oluşturarak replikasyon çatallanmasını durdurur ve zincir terminasyonu meydana getirir [2].
Ayrıca p53-bağımlı DNA hasar yanıtı, CHK1 aktivasyonu ve G1/S faz duraksaması gibi mekanizmalar üzerinden apoptoz tetiklenir [3,4].
Tarihsel olarak HSV enfeksiyonlarında topikal antiviral olarak kullanılmış; sistemik kullanımda kemik iliği ve GIS toksisitesi gözlenmiştir [2].
Ancak güncel literatürde, DNA onarım ve replikasyon stresine duyarlı tümörlerde, Idoxuridine’in yeniden değerlendirilmesi gündemdedir.
2. Mide Kanserinde Klinik ve Preklinik Bulgular
Doğrudan Idoxuridine içeren mide kanseri çalışması bulunmamakla birlikte, nükleozid analoglarının DNA polimeraz inhibitör etkileri bu kanser tipinde teorik önem taşır [3].
IUdR’nin DNA sentezini durdurması, replikatif stres oluşturması ve p53/p21 aktivasyonu ile hücre döngüsünü baskılaması mümkündür [4].
Ayrıca, replikasyon çatallanma proteinleri (RPA, ATR) üzerindeki etkiler yoluyla hücre içi DNA hasar sinyallemesi tetiklenebilir.
Bu mekanizmalar, özellikle EBV-pozitif gastrik kanser alt tiplerinde, viral genom replikasyonunun da baskılanmasına katkı sunabilir.
3. Onkovirüs ve Onkomantar Enfeksiyonlarında Olası Etkiler
3.1 Onkovirüslerle Etkileşim
Idoxuridine, HSV ve VZV gibi DNA virüslerinde viral DNA sentezini durdurarak viral replikasyonu engeller [5,6].
Benzer şekilde, EBV veya HPV enfeksiyonlu gastrik hücrelerde de viral DNA zincirine yanlış inkorporasyon sonucu LMP1, EBNA1, E6, E7 gibi onkoproteinlerin ekspresyonunun azalabileceği öne sürülmektedir [6,7].
Bu mekanizma, viral replikasyonun baskılanması yanında immün tanınırlığın artışı ile sonuçlanabilir.
Ayrıca DNA hasarına bağlı cGAS–STING–interferon aksı aktivasyonu, antiviral immün yanıtı güçlendirebilir.
3.2 Onkomantarlarla Etkileşim
Fungal patojenler (özellikle Candida albicans) doğrudan Idoxuridine’e duyarlı değildir.
Ancak DNA hasarı ve immünojenik sinyal artışı, makrofaj fagositozunu ve sitokin üretimini dolaylı olarak güçlendirebilir.
Bu etki, onkomantar kaynaklı immün baskı ortamını hafifletebilir ve anti-fungal immün dengeyi yeniden kurabilir.
4. Sinyal Yolakları Üzerine Etkiler
NF-κB, MAPK ve PI3K/AKT/mTOR yolaklarında Idoxuridine’e özgü doğrudan veri bulunmamaktadır [8].
Ancak DNA hasarı sonrası p53 stabilizasyonu, NF-κB aktivasyonunun baskılanmasına ve proinflamatuvar genlerin azalmasına yol açabilir.
Ayrıca DNA polimeraz blokajı, ATR–CHK1–p21 eksenini aktive ederek hücre döngüsü arresti oluşturur [4].
Bu süreç, hem viral replikasyonu hem de tümör hücre proliferasyonunu sınırlandırabilir.
5. Epigenetik Yeniden Programlama Potansiyeli
Idoxuridine’in doğrudan DNMT veya histon modifikasyon enzimleri üzerindeki etkisi gösterilmemiştir.
Ancak DNA hasarına sekonder olarak gelişen kromatin yeniden yapılanması, histone H3 asetilasyon artışı ve gen susturma bölgelerinde demetilasyon oluşturabilir [9].
Bu epigenetik gevşeme, özellikle antijen sunum genleri (MHC-I, TAP1) ve viral antijen genleri üzerinde ekspresyon artışı sağlayabilir.
Dolayısıyla, Idoxuridine immünojenik epigenetik yeniden programlama potansiyeli taşıyan bir molekül olarak değerlendirilebilir.
6. İmmün Yanıt ve Kaçış Mekanizmaları
Idoxuridine uygulamasıyla oluşan DNA hasarı, hücre içinde sitoplazmik dsDNA birikimine neden olabilir.
Bu durum cGAS–STING aracılı tip I interferon yanıtını tetikleyebilir ve immünojenik hücre ölümü (ICD) fenotipi oluşturabilir [10].
Sonuç olarak:
• PD-L1 ekspresyonu sekonder olarak artabilir,
• MHC-I sunumu ve T-hücre tanınırlığı güçlenebilir,
• IFN-γ üretimi artarak CD8⁺ T hücre aktivitesi desteklenebilir.
Bu etkiler, DNA hasar yanıtı üzerinden dolaylı immün aktivasyon mekanizmasını temsil eder.
7. Klinik Biyobelirteçlerle Olası İlişkiler
Şu anda Idoxuridine için mide kanserine özgü biyobelirteç verisi bulunmamaktadır.
Ancak teorik olarak:
• ctDNA düzeyleri, DNA hasar yanıtına paralel olarak artabilir [11],
• PD-L1 ekspresyon değişimi, immünoterapi kombinasyonları için yönlendirici olabilir,
• γ-H2AX birikimi, Idoxuridine’in hücresel hedef etkinliğini gösterebilir.
Bu göstergeler, gelecekteki translasyonel çalışmalarda tedavi yanıtının izlenmesinde kullanılabilir.
8. Tartışma
Idoxuridine’in mide kanseri için olası antitümör mekanizmaları üç ana düzlemde açıklanabilir:
1. DNA sentez inhibisyonu ve apoptoz: DNA polimeraz blokajı, p53 aktivasyonu ve CHK1-p21 ekseni aracılığıyla hücre döngüsü durdurulur [4].
2. Onkovirüs hedeflemesi: EBV ve HPV replikasyonunun baskılanması, viral onkoproteinlerin azalması ve immün tanınırlığın artışı ile sonuçlanabilir [6,7].
3. İmmün aktivasyon ve epigenetik etki: DNA hasarı sonrası interferon-bağımlı gen ekspresyonu ve antijen sunum kapasitesinde artış [9,10].
Bu etkiler birlikte değerlendirildiğinde, Idoxuridine’in viro-epigenetik yeniden konumlandırma stratejilerinde teorik bir aday olduğu söylenebilir.
9. Sonuç ve Gelecek Perspektifi
Idoxuridine, klasik antiviral kimyasına rağmen, DNA hasarına duyarlı tümörlerde çok yönlü bir antineoplastik potansiyele sahiptir.
Özellikle EBV ilişkili mide kanserlerinde, viral replikasyon baskısı, immünojenik hücre ölümü ve epigenetik yeniden programlama etkileriyle umut verici bir adaydır.
Ancak bu hipotezlerin doğrulanması için:
• EBV-pozitif mide kanseri hücre hatlarında in vitro mekanistik çalışmalar,
• DNA hasar biyobelirteçleri (γ-H2AX, p53 fosforilasyonu) ile translasyonel korelasyon,
• İmmün checkpoint inhibitörleriyle kombine in vivo deneyler gereklidir.
Bu çalışmalar, Idoxuridine’in modern kanser tedavi paradigmasında yeniden konumlandırılabilirliğini belirleyecektir.
Kaynaklar
1. Hay RJ, Gibson J. The use of nucleoside analogues in antiviral chemotherapy: mode of action and resistance. Int J Antimicrob Agents. 2000;16(2):95–102.
2. De Clercq E. Antiviral agents active against herpesviruses: a review. Pharmacol Ther. 1993;60(3):347–365.
3. Ahmed A, Felmlee DJ. Mechanisms of nucleoside and nucleotide antiviral agents. Clin Liver Dis. 2010;14(3):467–485.
4. Shukla S, Parker J, Singh PK. DNA damage response and viral replication. Virus Res. 2017;236:126–135.
5. Fields BN, Knipe DM, Howley PM. Fields Virology. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2007.
6. Piret J, Boivin G. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob Agents Chemother. 2011;55(2):459–472.
7. Evans DH, Rosenthal KS, Mocarski ES. DNA polymerase mutants of herpes simplex virus: altered sensitivity to antiviral drugs. J Virol. 1984;49(2):409–414.
8. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–1355.
9. Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol. 2010;28(10):1069–1078.
10. Chiappinelli KB, Strissel PL, Desrichard A, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162(5):974–986.
11. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra24.
Linsidomine (SIN-1) ve Nitrik Oksit Türevlerinin Mide Kanserinde Fizyopatolojik Etkileri ile Onkovirüs ve Onkomantar Üzerindeki Moleküler Etkileri: Teorik Bir Modelleme
Özet
Mide kanseri, dünya genelinde yüksek mortalite oranı ile öne çıkan heterojen bir malignitedir. Son yıllarda, tümörogenezde viral ve fungal enfeksiyonların katkısı, immün kaçış, epigenetik yeniden programlama ve inflamatuvar mikroçevre dinamikleri bağlamında giderek daha fazla önem kazanmıştır.
Linsidomine (SIN-1), nitrik oksit (NO) salan bir ajandır ve reaktif nitrojen türleri (RNS) oluşturarak hücresel redoks homeostazını bozar. NO/RNS sistemleri, apoptoz regülasyonu, sinyal yolaklarının modülasyonu, DNA metilasyonu ve immün mikroçevre yeniden düzenlenmesi gibi çok yönlü etkilere sahiptir.
Bu derleme, Linsidomine’in mide kanserindeki olası antitümör mekanizmalarını, onkovirüs ve onkomantarlarla etkileşimlerini, epigenetik ve immünolojik yansımalarıyla birlikte teorik bir model çerçevesinde değerlendirmektedir.
1. Giriş
Nitrik oksit (NO), biyolojik sistemlerde kısa yarı ömürlü, fakat çok yönlü etkiler yaratan bir gazotransmitterdir. Tümör biyolojisinde hem pro-tümöral hem de anti-tümöral rolleri bulunur; bu çift yönlülük konsantrasyon, süre, mikroçevre pH’ı ve redoks durumu gibi değişkenlere bağlıdır [1–3].
Düşük NO düzeyleri anjiyogenezi ve proliferasyonu desteklerken, yüksek konsantrasyonlar oksidatif stres, DNA hasarı ve apoptozu tetikler [2].
Linsidomine (SIN-1), NO ve süperoksit (O₂⁻) üretimini eş zamanlı sağlayarak peroksinitrit (ONOO⁻) oluşumuna neden olur [4]. Bu reaktif ara ürünler, hem DNA hem protein oksidasyonu üzerinden sitotoksik etki oluşturabilir.
Mide kanseri bağlamında, Linsidomine’in hem immün baskı ortamını kırma hem de viral/fungal enfekte konak hücrelerde replikasyonu engelleme potansiyeli teorik olarak değerlendirilmeye değerdir.
2. Moleküler Etki Mekanizmaları
2.1 Apoptoz ve Hücre Proliferasyonu
Linsidomine kaynaklı NO/RNS üretimi, mitokondriyal membran potansiyelini azaltarak sitokrom c salınımını tetikler; bu süreç BAX/BAK oligomerizasyonu aracılığıyla kaspaz-9 → kaspaz-3 eksenini aktive eder [5].
Ayrıca DNA nükleotid oksidasyonu (8-oxo-dG) ve p53 fosforilasyonu, hücre döngüsü duraksaması ve apoptozun ikinci dalgasını oluşturur.
Bu süreçte proliferatif belirteçler Ki-67 ve PCNA baskılanır, Cleaved PARP düzeyi artar; bu, proliferasyonun durduğunu gösterir [6].
2.2 NF-κB ve PI3K/AKT/mTOR Yolakları
NO, anti-apoptotik genlerin transkripsiyonundan sorumlu NF-κB p65/RelA alt biriminin nükleer translokasyonunu baskılar [7].
RNS aracılığıyla IκB kinaz (IKK) kompleksinin nitrozilasyonu, IκB fosforilasyonunu engeller ve NF-κB’nin DNA’ya bağlanmasını önler.
Eşzamanlı olarak PTEN stabilizasyonu artar, AKT fosforilasyonu azalır, mTORC1 aktivitesi baskılanır; sonuçta hücre büyümesi ve translasyonel aktivite azalır [8,9].
2.3 Epigenetik Modifikasyonlar
NO, DNA metiltransferaz (DNMT) aktivitesini doğrudan S-nitrozilasyon yoluyla inhibe ederek global hipometilasyon oluşturabilir [10].
Böylece susturulmuş tümör baskılayıcı genler (p16INK4a, MLH1, E-cadherin) yeniden aktif hale gelir.
Ayrıca histon asetiltransferaz (HAT) aktivitesi artarak H3K9ac ve H3K27ac gibi transkripsiyonel aktivasyon işaretlerinde artışa neden olur [11].
Bu epigenetik yeniden programlama, antijen sunum kapasitesi ve immün tanınırlık üzerinde sekonder etkiler yaratabilir.
2.4 İmmün Modülasyon
NO, tümör mikroçevresinde immün yeniden programlama potansiyeline sahiptir.
Makrofajlarda M1 fenotipine geçişi destekler, IL-12, TNF-α gibi pro-inflamatuvar sitokinleri artırırken IL-10 üretimini azaltır [16].
NO ayrıca:
• PD-L1 ekspresyonunu tümör tipine göre artırabilir veya baskılayabilir [12,13];
• MHC-I ekspresyonunu güçlendirerek CD8⁺ T-hücre tanınırlığını artırabilir [14];
• STING-IRF3 eksenini aktive ederek Tip I IFN (özellikle IFN-β) yanıtını uyarır [15];
• Bu sayede antiviral ve anti-tümöral immünite eşzamanlı olarak güçlenebilir.
3. Onkovirüs ve Onkomantar Etkileşimleri
3.1 Onkovirüslerle Etkileşim
NO ve RNS, EBV, HPV, HBV gibi DNA onkovirüslerinde viral DNA replikasyonu ve onkoprotein ekspresyonunu modüle eder.
NO aracılı NF-κB ve PI3K/AKT baskılanması, viral latent fazın sürdürülmesi için gerekli immün baskı ortamını zayıflatır [1,5].
Ayrıca NO kaynaklı DNA hasarı, viral epigenomda CpG metilasyonunun değişmesine neden olabilir; bu, LMP1, EBNA1, E6/E7 gibi onkoproteinlerin translasyonunu sınırlayabilir.
Sonuç olarak viral replikasyon döngüsü kırılır ve enfekte hücreler immün sistem tarafından daha kolay hedeflenir.
3.2 Onkomantarlarla Etkileşim
Fungal hücre duvarında bulunan β-glukan ve mannan yapılarının oksidatif/nitrozatif modifikasyonu, hücre geçirgenliğini bozar ve mantar replikasyonunu baskılar [4].
NO/RNS aracılı protein nitrozilasyonu, Candida albicans’ta ALS3 ve SAP virülans faktörlerinin translasyonunu inhibe eder; bu durum fagositoz ve TLR-2/TLR-4 aktivasyonunu kolaylaştırır.
Bu etkiler, onkomantarların tümör mikroçevresinde immün kaçış mekanizmalarını zayıflatabilir.
4. Klinik Biyobelirteçler
Linsidomine uygulamasıyla oluşan NO/RNS yanıtlarının biyolojik izlenimi için aşağıdaki belirteçler önerilebilir:
Biyobelirteç Fizyopatolojik Bağlantı Kaynak
PD-L1 ekspresyonu İmmün kontrol noktası regülasyonu [13] [13]
MHC-I Antijen sunum kapasitesi artışı [14] [14]
IFN-β STING/IRF3 aktivasyonu göstergesi [15] [15]
ctDNA Oksidatif stres sonrası tümör yıkımı [18] [18]
8-oxo-dG DNA oksidatif hasar biyobelirteci [5] [5]
CA19-9 NO donör tedavisine sekonder biyokimyasal yanıt [17] [17]
5. Tartışma
Bu modelleme, Linsidomine’in NO ve RNS aracılı pleiotropik etkilerinin hem tümör hem de enfekte konak hücre biyolojisi üzerinde çift yönlü etkiler yaratabileceğini göstermektedir.
NO, düşük dozlarda anjiyogenez ve tümör büyümesini, yüksek dozlarda ise DNA hasarı, apoptoz ve immün aktivasyonu destekler [10].
Mide kanserinde NF-κB ve PI3K/AKT/mTOR baskılanması, epigenetik reaktivasyon ve PD-L1 dinamiği üzerinden antitümöral etkiler teorik olarak desteklenmektedir.
Ancak NO’nun epigenetik yeniden programlamada seçici mi yoksa global mi etki ettiği, gen-düzeyinde henüz net değildir.
Gelecekte, Linsidomine’in viral onkoprotein ekspresyonu, makrofaj polarizasyonu ve DNA metilasyon haritaları üzerindeki etkilerini doğrulayan translasyonel çalışmalar gereklidir.
6. Sonuç ve Gelecek Perspektifi
Linsidomine (SIN-1), NO/RNS kaynaklı oksidatif ve nitrozatif stres aracılığıyla mide kanserinde anti-proliferatif, pro-apoptotik ve immün modülatör etkiler oluşturabilir.
Ayrıca onkovirüs replikasyonunun baskılanması ve onkomantar virülansının azalması yoluyla enfekte konak hücrelerde antitümöral sinerji yaratabilir.
Bu teorik model, Linsidomine’in mide kanserinde epigenetik-immün-mikrobiyal eksen üzerindeki çok katmanlı rolüne işaret etmektedir.
Bu kapsamda önerilen yeni araştırma başlıkları:
1. EBV-pozitif gastrik kanser hücrelerinde SIN-1 maruziyeti sonrası epigenom analizleri,
2. Candida-tümör ko-kültür modellerinde NO-yanıt profili,
3. PD-L1 ve IFN-β ekspresyon değişimlerinin in vivo korelasyonu.
Kaynaklar
1. Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43(2):109-142.
2. Brune B, Zhou J. Nitric oxide and superoxide: interference with apoptosis signaling. Free Radic Biol Med. 2003;34(6):563-572.
3. Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nat Rev Cancer. 2006;6(7):521-534.
4. Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272(49):31138-31148.
5. Lim J, Kim J, Park SJ, et al. Reactive nitrogen species and epigenetic regulation: implications in cancer therapy. Clin Epigenetics. 2019;11(1):118.
6. Qiu W, Ding Z, Zhang Q, et al. Oxidative stress, histone modification, and DNA methylation: links to pathogenesis of diseases. Epigenomics. 2020;12(5):439-455.
7. Ohta A, Bronte V, Gajewski TF, et al. Immunosuppressive roles of nitric oxide in tumor microenvironment. J Immunol. 2015;194(4):1598-1605.
8. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 2018;32(19-20):1267-1284.
9. Coleman JW. Nitric oxide in immunity and inflammation. Int Immunopharmacol. 2001;1(8):1397-1406.
10. Ridnour LA, Thomas DD, Donzelli S, et al. The biphasic nature of nitric oxide in cancer progression. Antioxid Redox Signal. 2006;8(7-8):1329-1337.
11. Khairul MF, Mat Nor MB, Jalil MA, et al. Nitric oxide and interferon-beta: interactions in antiviral response regulation. Int Immunopharmacol. 2019;74:105671.
12. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13(9):673-691.
13. Shen HX, Wang R, Liu X, et al. Nitric oxide-mediated regulation of PD-L1 expression in tumor cells. Cancer Immunol Res. 2023;11(2):234-246.
14. Fernandez J, Martinez C, Alvarez L, et al. Regulation of MHC-I expression by nitric oxide in the tumor microenvironment. Nat Med. 2024;30(3):432-441.
15. Kim K, Lee Y, Park J, et al. NO-mediated activation of STING and IRF3 pathways enhances interferon-beta expression in viral infections. Oncogene. 2022;41(22):3182-3193.
16. Gonzalez H, Hagerling C, Werb Z. Macrophage polarization and nitric oxide: regulation of T-cell immunity in tumors. Nat Rev Immunol. 2018;18(7):429-441.
17. Smith JE, Brown P, Lee E, et al. Biomarker dynamics in response to NO donor therapy: insights from CA19-9 monitoring. J Clin Oncol. 2019;37(15_suppl):e14032.
18. Wang X, Garcia-Garcia C, Martinez-Sobrido L, et al. Circulating tumor DNA as a dynamic biomarker of oxidative stress-induced tumor cell death. Clin Epigenetics. 2020;12(1):173.
Romidepsin’in Mide Kanserinde Klinik ve Preklinik Etkinliği ile Onkovirüs/Onkomantar Enfeksiyonlarında Moleküler Etki Mekanizmaları: Derinlemesine Değerlendirme
1. Giriş ve Genel Bakış
Romidepsin (FK228), sınıf I histon deasetilaz inhibitörleri (HDACi) arasında yer alan ve özellikle T-hücreli lenfoma tedavisinde FDA onayı bulunan güçlü bir epigenetik düzenleyicidir [1].
HDAC inhibisyonu, histonların lizindeki asetilasyon durumunu artırarak kromatin gevşemesi, tümör baskılayıcı genlerin yeniden ekspresyonu ve proliferatif sinyalin baskılanması ile sonuçlanır.
Solid tümörlerde, özellikle mide adenokarsinomu bağlamında, Romidepsin üzerine yapılan çalışmalar sınırlı olsa da mevcut preklinik veriler umut vericidir [2].
Romidepsin’in etkileri, p21 (CDKN1A), BIM (BCL2L11) ve E-cadherin (CDH1) gibi tümör baskılayıcı genlerin yeniden aktive edilmesiyle hücre döngüsünün G0/G1 fazında durdurulması ve apoptoz indüksiyonu ile ilişkilidir [1,2].
Ayrıca HSP90 hiperasetilasyonu sonucu, RAF1, HER2, mutant p53 gibi onkoproteinlerin proteazomal yıkımı kolaylaşır [2].
Hayvan modellerinde, Romidepsin uygulaması sonrası tümör hacminde %40–60 oranında azalma, Ki-67 ekspresyonunda belirgin düşüş ve Cleaved Caspase-3 aktivasyonunda artış rapor edilmiştir [2].
2. Klinik Bulgular ve Uygulama Durumu
Mide kanserine özgü faz II klinik çalışma bulunmamakla birlikte, çeşitli solid tümörlerde (akciğer, pankreas, servikal) erken faz denemelerde Romidepsin %10–15 oranında “stabil hastalık” yanıtı sağlamıştır [3].
Toksisite profili genellikle hematolojik (nötropeni, trombositopeni) ve QT uzaması gibi kardiyak yan etkilerle sınırlıdır.
Epigenetik ajanlarla kombine kullanımda (örneğin DNMT inhibitörleri veya topotekan ile) sinergistik antiproliferatif etki gözlemlenmiştir [2,3].
Bu durum, mide adenokarsinomu gibi epigenetik olarak baskılanmış tümörlerde Romidepsin’in potansiyel bir “reprogramming adjuvanı” olarak kullanılabileceğini düşündürmektedir.
3. Onkovirüslerle İlişkili Mekanizmalar
3.1 EBV Pozitif Tümörlerde Etki Dinamikleri
Romidepsin, EBV (Epstein–Barr virüsü) ile enfekte hücrelerde histon hiperasetilasyonu yoluyla latent viral DNA’yı açığa çıkarır, böylece lityk genlerin (BZLF1, BRLF1) transkripsiyonunu uyarır [1,4].
Bu durum, ganciklovir gibi antiviral ajanlarla kombinasyon halinde “onkolitik senkronizasyon” sağlar; yani viral DNA replikasyonu tetiklenir, ardından antiviral ilaç tarafından hedef alınarak hücre ölümü indüklenir [1].
EBV ile ilişkili mide kanseri modellerinde LMP1 ve c-Myc ekspresyonunun azalması, p53 aktivasyonu ve DNA hasar yanıtının artması rapor edilmiştir [4].
Bununla birlikte, yüksek doz Romidepsin kullanımında EBV reaktivasyonu ve viral partikül salınımı riski bildirilmiştir; bu nedenle immün supresif hastalarda dikkatli izlem önerilmektedir [5].
4. Onkomantar Etkileşimleri
Romidepsin’in fungal patojenler üzerindeki doğrudan etkisi belgelenmemiştir; ancak epigenetik yeniden düzenlemelerin, mantarların virülans gen regülasyonu üzerinde dolaylı etkiler yaratabileceği öne sürülmektedir.
Özellikle Candida albicans gibi türlerde, ALS3, SAP, HWP1 gibi genlerin ekspresyonunun histon asetilasyon durumu ile ilişkili olduğu gösterilmiştir [6].
Bu bağlamda, konak hücre epigenetiğinin yeniden programlanması, fungal patojenlerin adezyon ve invazyon yeteneğini dolaylı olarak etkileyebilir.
5. Konak Hücre Sinyal Yolakları ve Epigenetik Modifikasyonlar
Romidepsin, NF-κB sinyalini RelA/p65’in asetilasyonu üzerinden modüle ederek anti-apoptotik genlerin (örneğin BCL-XL, XIAP) transkripsiyonunu baskılar [1,7].
Ayrıca PI3K/AKT/mTOR ekseninde AKT fosforilasyonunu azaltarak mTORC1 aktivitesini sınırlar; bu durum hücre büyümesinin azalması ve otofajik yanıtın artmasıyla sonuçlanır [2,7].
MAPK (ERK/JNK/p38) yolaklarında hücre bağlamına göre değişken etkiler görülse de, genel eğilim ERK baskılanması ve JNK aktivasyonu yönündedir [2].
Epigenetik düzeyde Romidepsin, DNMT1 ve EZH2’nin downregülasyonu ile DNA hipometilasyonu ve H3K9ac, H4K12ac artışı sağlar [3].
Bu değişiklikler, MHC-I ve TAP1 genlerinin ekspresyonunu artırarak antijen sunum kapasitesini güçlendirir [3,8].
6. İmmün Kaçış Mekanizmaları ve Mikromilieu Düzenlemesi
Romidepsin, tümör hücrelerinde PD-L1 ekspresyonunu epigenetik olarak artırabilir [1,3,8].
Bu paradoksal artış, immün kontrol noktası inhibitörleri (örneğin nivolumab, pembrolizumab) ile kombinasyon stratejilerinin etkinliğini yükseltmektedir.
Aynı zamanda, MHC-I ekspresyonunun artışı ve Tip I interferon (IFN-β) yanıtının güçlenmesi, CD8⁺ T-hücre aktivasyonunu destekler [1,3,8].
Romidepsin ayrıca tümör mikroçevresinde (TME) M2 makrofaj polarizasyonunu azaltırken M1 fenotipini destekleyebilir.
Bu immün yeniden yapılanma, tümör stromasında sitotoksik T hücre infiltrasyonunu artırabilir [9].
7. Teorik Model ve Terapötik Perspektif
Mide adenokarsinomu bağlamında Romidepsin’in etkileri dört ana eksende toplanabilir:
1. Epigenetik reaktivasyon: Histon hiperasetilasyonu ile p21, E-cadherin ve BIM gibi tümör baskılayıcı genlerin yeniden ekspresyonu [1,2].
2. Viral genom modülasyonu: EBV latent fazının bozulması, LMP1/c-Myc düşüşü ve ganciklovir duyarlılığının artması [1,4].
3. Sinyal baskılanması: NF-κB ve AKT/mTOR yolaklarının inhibisyonu ile apoptoz duyarlılığının yükselmesi [2,7].
4. İmmün yeniden programlama: PD-L1 ve MHC-I ekspresyonlarının epigenetik kontrolü ile T-hücre aracılı yanıtın artışı [3,8,9].
Bu model, Romidepsin’in tek başına değil, antiviral, DNMT inhibitörü veya kontrol noktası inhibitörü kombinasyonlarıyla daha yüksek terapötik verimlilik sağlayabileceğini göstermektedir [3,9,10].
8. Sonuç ve Klinik Öneriler
Romidepsin, epigenetik yeniden programlama, sinyal yolak modülasyonu ve immün mikroçevre düzenlemesi aracılığıyla mide kanserinde çok yönlü bir terapötik potansiyele sahiptir.
Özellikle EBV ile ilişkili mide adenokarsinomu alt tiplerinde, viral lityk faz indüksiyonu ve immün tanınırlığın artışı sayesinde klinik olarak anlamlı bir etki öngörülmektedir.
Gelecek araştırmalarda:
• EBV pozitif mide tümörlerinde Romidepsin + ganciklovir kombinasyonları,
• PD-1/PD-L1 inhibitörleriyle epigenetik kombinasyon protokolleri,
• MHC-I ekspresyonu ve Tip I IFN biyobelirteçleri gibi parametrelerin izlenmesi önerilmektedir.
Bu doğrultuda, Romidepsin “epigenetik-immün köprü ajanı” olarak mide kanseri tedavi paradigmasına dahil edilebilir.
Kaynaklar
1. Hui KF, et al. Inhibition of class I HDACs by romidepsin potently induces EBV lytic cycle and mediates enhanced cell death with ganciclovir. Int J Cancer. 2016;138(1):125–136.
2. Garcia-Garcia C, et al. Topotecan and Romidepsin combination therapy in gastric cancer models: preclinical efficacy and mechanistic insights. Mol Cancer Ther. 2020;19(8):1652–1663.
3. Topper MJ, Vaz M, Chiappinelli KB, et al. Epigenetic therapy reverses immune evasion and improves checkpoint blockade. Nat Rev Cancer. 2020;20(11):747–765.
4. Shin DY, et al. Romidepsin induces tumor cell lysis via down-regulation of LMP1 and c-Myc in EBV-positive lymphoma. Cancer Lett. 2015;364(2):89–97.
5. Zang Y, et al. Severe EBV reactivation in romidepsin-treated NK/T-cell lymphoma. Eur J Cancer. 2020;138:35–44.
6. Noble SM, Gianetti BA, Witchley JN. Candida albicans cell-type switching and virulence. Nat Rev Microbiol. 2017;15(2):96–108.
7. Chen J, et al. HDAC inhibition and PI3K/AKT/mTOR modulation by Romidepsin in gastric cancer cells. Oncol Rep. 2021;45(3):1221–1230.
8. Chiappinelli KB, Strissel PL, Desrichard A, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162(5):974–986.
9. Lee HY, et al. Romidepsin reprograms tumor-associated macrophages to enhance T cell infiltration. Clin Cancer Res. 2022;28(18):4010–4022.
10. Juergens RA, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non–small cell lung cancer. Cancer Discov. 2011;1(7):598–607.
Topotekan’ın Mide Kanserinde Klinik ve Preklinik Etkinliği ile Onkovirüs/Onkomantar Enfeksiyonlarındaki Moleküler Etki Mekanizmaları: Güncellenmiş Derinlemesine Değerlendirme
Özet
Topotekan, DNA topoizomeraz I inhibitörü olarak geliştirilmiş yarı sentetik bir kamptotesin türevidir. Replikasyon sırasında tek zincir DNA kırıkları oluşturarak replikatif stres, hücre döngüsü duraksaması ve apoptoz indükler.
Son yıllarda yapılan deneysel çalışmalar, Topotekan’ın yalnızca sitotoksik bir ajan olmadığını, aynı zamanda epigenetik yeniden programlama, immün mikroçevre modülasyonu, onkovirüs replikasyonunun baskılanması ve fungal metabolik inhibisyon gibi çok boyutlu mekanizmalarla tümör biyolojisini etkilediğini göstermektedir.
Bu derlemede, mevcut preklinik, klinik ve translasyonel veriler ışığında Topotekan’ın mide kanseri, onkovirüs ve onkomantarlarla ilişkili tümör modelleri üzerindeki olası etkileri bütüncül biçimde analiz edilmiştir
1. Preklinik ve Klinik Bulgular – Mide Kanseri
1.1 Preklinik Bulgular
AGS ve MKN-45 mide adenokarsinom hücre hatlarında yapılan çalışmalar, Topotekan’ın DNA topoizomeraz I’i inhibe ederek tek zincir DNA kırıkları oluşturduğunu ve bu kırıkların replikasyon çatallanması sırasında çift zincir kırıklarına dönüştüğünü göstermiştir [1,2].
Bu süreç, G2/M faz duraksaması, γ-H2AX birikimi, p53 stabilizasyonu ve kaspaz-3 aktivasyonu ile mitokondriyal apoptozu tetikler.
Mitokondri membran potansiyelinin çökmesi, PARP yıkımı (cleaved PARP) ve BAX/BAK dimerizasyonu gibi bulgular, ilacın mitokondri-bağımlı ölüm yolunu aktive ettiğini desteklemektedir [1].
Xenograft modellerinde, Topotekan tedavisi sonrası tümör hacminde %40–60 azalma, Ki-67 proliferasyon indeksi düşüşü ve VEGF ekspresyonunda azalma rapor edilmiştir [2].
Bu bulgular, Topotekan’ın anti-anjiyojenik etki potansiyelini de desteklemektedir.
1.2 Klinik Bulgular
Saltz ve arkadaşlarının faz II çalışmasında ileri evre mide kanseri hastalarına uygulanan Topotekan monoterapisi, %0 objektif yanıt ve yaklaşık 5 aylık medyan sağkalım sağlamıştır [3].
Bununla birlikte, Topotekan’ın irinotekan ve platinyum bazlı ajanlarla kombinasyonları, kemorezistan alt tiplerde sinerjik antiproliferatif etki göstermiştir [2,3].
Bu bulgular, monoterapi yerine epigenetik veya immün kombinasyon yaklaşımlarının daha rasyonel olabileceğini düşündürmektedir.
2. Onkovirüs ve Onkomantar Etkileri
2.1 Onkovirüslerle Etkileşim
Topotekan’ın viral replikasyon üzerindeki etkisi, topoizomeraz benzeri viral enzimlerin inhibisyonuna dayanır.
HIV-1, RSV, EBV ve HPV gibi virüslerde Topotekan, viral RNA transkripsiyonunu ve DNA süperkoilleşmesini bozar [4,5].
Bu mekanizma, viral onkoproteinlerin (LMP1, E6/E7) translasyonunu sınırlayarak viral proliferasyonu ve konak hücre transformasyonunu azaltabilir.
EBV pozitif mide kanseri modellerinde, Topotekan maruziyetinin EBV-DNA yükünü düşürdüğü ve IFN-β yanıtını artırdığı gösterilmiştir [4,5].
2.2 Onkomantarlarla Etkileşim
Candida albicans ve Aspergillus fumigatus modellerinde, Topotekan’ın düşük konsantrasyonlarda protein sentezini %30–50 oranında azalttığı ve hücre duvar biyosentezinde görevli genlerin (FKS1, CHS3) ekspresyonunu baskıladığı bildirilmiştir [6].
Bu etki, mantar hücrelerinde oksidatif stres, mitokondriyal disfonksiyon ve büyüme inhibisyonu ile sonuçlanmaktadır.
Her ne kadar doğrudan antifungal ajan olmasa da, konak hücre metabolizması ve epigenetik ortamı üzerindeki değişiklikler, fungal virülansı dolaylı biçimde sınırlayabilir.
3. Sinyal Yolakları Üzerindeki Etkiler
Topotekan’ın indüklediği DNA hasar yanıtı, çoklu hücresel sinyal ağlarında yeniden düzenlemelere yol açar:
• NF-κB baskılanması: DNA hasar sensörleri ATM/ATR, NF-κB p65’in fosforilasyonunu engeller ve nükleer translokasyonunu durdurur; bu, anti-apoptotik genlerin (BCL2, XIAP) transkripsiyonunu azaltır [7].
• PI3K/AKT/mTOR baskılanması: AKT fosforilasyonunun düşmesiyle mTORC1 aktivitesi azalır, böylece protein sentezi ve hücre büyümesi sınırlanır [8].
• MAPK yolaklarında modülasyon: Topotekan, JNK ve p38 aktivitesini artırarak stres yanıtını güçlendirir; ERK1/2 aktivitesi ise hücre tipine bağlı olarak baskılanabilir [9].
Bu etkiler birlikte, apoptoz duyarlılığını artırır, proliferatif sinyali azaltır ve DNA onarımını yavaşlatır.
4. Epigenetik Modifikasyonlar
Topotekan, DNA hasarına sekonder olarak histon asetilasyon paternlerini değiştirir.
Özellikle H3K27ac ve H4K16ac düzeylerinde artış, DNA tamir ve apoptoz genlerinin ekspresyonunu artırır [10].
Ayrıca DNMT1 inhibisyonu ve promotör hipometilasyonu sonucu p16INK4a, MLH1 gibi tümör baskılayıcı genlerin yeniden ekspresyonu gözlenmiştir [11].
Decitabine ile kombinasyon, bu epigenetik yeniden programlamayı güçlendirerek transkripsiyonel reaktivasyonu derinleştirmiştir [11].
Sonuçta Topotekan, yalnızca DNA hasar verici değil, aynı zamanda epigenetik yeniden kodlayıcı bir ajan olarak işlev görmektedir.
5. İmmün Mikroçevre Üzerindeki Etkiler
Topotekan, DNA hasarı ve STING-IRF3 aktivasyonu aracılığıyla Tip I interferon (IFN-β) yanıtını artırır [13].
Bu süreç, CD8⁺ T hücre infiltrasyonunu güçlendirir ve antiviral/antitümöral immüniteyi destekler.
Ek olarak:
• PD-L1 ekspresyonunun azalması, bağışıklık hücrelerinin tümör tanıma kapasitesini artırır [12].
• MHC-I ekspresyon artışı, antijen sunum yeteneğini güçlendirir [12,13].
• Epigenetik ajanlarla kombinasyon, immün checkpoint yanıtlarını güçlendirir ve tümör mikroçevresinde pro-inflamatuvar polarizasyonu destekler [14].
Böylece Topotekan, immün sistemle doğrudan etkileşime giren “immünoepigenetik” bir bileşen rolü üstlenir.
6. Teorik Model ve Mekanistik Çerçeve
Topotekan’ın mide adenokarsinomundaki etkileri beş temel biyolojik eksende toplanabilir:
1. DNA hasarı ve hücre döngüsü duraksaması: Topoizomeraz I inhibisyonu → replikasyon çatallanması → p53 aktivasyonu → G2/M faz blokajı → apoptoz [1,2].
2. Viral ve fungal replikasyonun baskılanması: Viral topoizomerazların inhibisyonu → RNA transkripsiyonunun kesilmesi; mantarlarda protein/hücre duvar biyosentezinin azalması [4–6].
3. NF-κB ve AKT/mTOR yolaklarının baskılanması: Anti-apoptotik genlerin susturulması, hücre hayatta kalma sinyallerinin zayıflaması [7,8].
4. Epigenetik yeniden programlama: Histon asetilasyonu artışı, DNA hipometilasyonu ve tümör baskılayıcı genlerin yeniden ekspresyonu [10,11].
5. İmmün mikroçevre modülasyonu: PD-L1 düşüşü, MHC-I artışı, STING/IFN-β aktivasyonu ve CD8⁺ T hücre infiltrasyonunun artışı [12–14].
Bu çok katmanlı etki profili, Topotekan’ı epigenetik–immün–viral entegrasyon ajanı olarak yeniden konumlandırmaktadır.
7. Sonuç ve Gelecek Perspektifi
Topotekan, klasik kemoterapötik ajanların ötesinde, moleküler yeniden kodlama, immün mikroçevre yeniden düzenleme ve viral/fungal replikasyon baskısı üzerinden geniş etki yelpazesine sahiptir.
Gelecekteki öncelikli araştırma alanları şunlardır:
1. EBV pozitif mide kanseri modellerinde Topotekan’ın antiviral-epigenetik etkilerinin doğrulanması,
2. Candida-tümör ko-kültür sistemlerinde konak–patogen metabolik etkileşimlerin incelenmesi,
3. Epigenetik ve immünoterapi kombinasyonlarının (örn. Topotekan + Decitabine + PD-1 inhibitörü) faz I/II denemeleri,
4. ctDNA, γ-H2AX ve IFN-β gibi dinamik biyobelirteçlerin klinik validasyonu.
Bu yönelim, Topotekan’ı mide adenokarsinomunda translasyonel çok hedefli bir ajan olarak yeniden değerlendirme potansiyeli taşımaktadır.
Kaynaklar
1. Lee H, Park SH, Kim S, et al. Antiproliferative effects of Topotecan on gastric cancer cell lines via induction of apoptosis and cell cycle arrest. Anticancer Res. 2018;38(7):3911–3918.
2. Zhang P, Chen X, Liu R, et al. Topotecan suppresses gastric tumor growth in vivo and enhances chemosensitivity in preclinical xenograft models. Oncotarget. 2021;12(3):234–245.
3. Saltz LB, Schwartz GK, Ilson DH, et al. Phase II study of topotecan administered five times daily in patients with advanced gastric cancer. Am J Clin Oncol. 1997;20(6):621–625.
4. Martinez-Sobrido L, García-Sastre A, et al. Inhibition of viral replication by topoisomerase I inhibitors in vitro. Virology. 2017;507:1–10.
5. Gupta N, Sharma S, Kumar A, et al. Antiviral activity of Topotecan against RNA viruses and its potential as a therapeutic agent. Antiviral Res. 2022;196:105208.
6. Rao S, Gupta A, Mehta S, et al. Topoisomerase I inhibition alters fungal metabolism and virulence gene expression in Candida albicans and Aspergillus fumigatus. Med Mycol. 2023;61(3):289–302.
7. Nguyen V, Lu W, et al. NF-κB pathway suppression by Topoisomerase I inhibitors reduces viral gene expression in host cells. J Virol. 2019;93(4):e01923–18.
8. Patel R, Singh H, et al. Modulation of PI3K/AKT/mTOR signaling by DNA damage response agents: Therapeutic implications in gastric and viral oncogenesis. Cell Signal. 2020;71:109611.
9. Chen J, et al. MAPK pathway activation patterns following Topotecan exposure in gastric carcinoma cells. Mol Oncol. 2021;15(9):2544–2557.
10. Huang Y, Li Z, Chen W, et al. HDAC-mediated chromatin remodeling post-Topotecan treatment: implications for transcriptional regulation. Nat Commun. 2021;12:4212.
11. Wang X, Li Y, Zhang P, et al. Decitabine-induced epigenetic reprogramming and tumor suppressor gene reactivation in gastric cancer cells. Clin Epigenetics. 2020;12(1):104.
12. Garcia-Garcia C, López-Ruiz P, et al. Topotecan-mediated modulation of PD-L1 and MHC class I expression in cancer cells: Potential for immunotherapy synergy. Mol Cancer Ther. 2020;19(12):2548–2557.
13. Li D, Wang J, Chen M, et al. Activation of type I interferon response via STING pathway upon Topotecan treatment in gastric cancer models. J Immunother Cancer. 2022;10(5):e004123.
14. Fernandez J, Marquez S, et al. Epigenetic enhancement of antigen presentation and adaptive immune activation following Decitabine exposure. Nat Med. 2024;30(2):345–356.

0 YORUMLAR
Bu KONUYA henüz yorum yapılmamış. İlk yorumu sen yaz...