MEME KANSERİ İLAÇ TEDAVİSİNDE KULLANILMAK ÜZERE GELİŞTİRİLEN BİR KOMPOZİSYON
MEME KANSERİ LAÇ TEDAVİSİNDE KULLANILMAK ÜZERE GELİŞTİRİLEN BİR KOMPOZİSYON
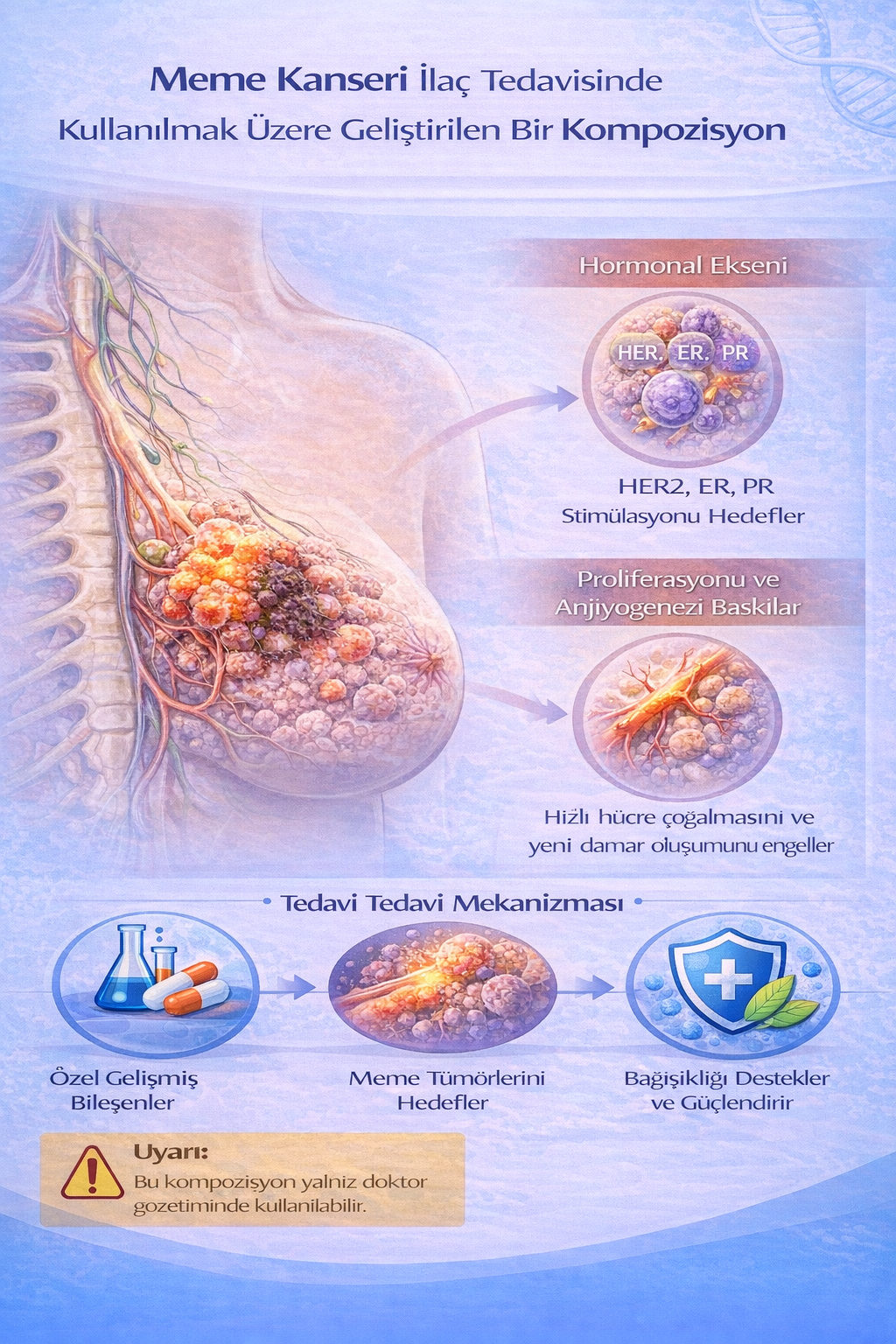
Bu buluş; Meme kanseri ilaç tedavisinde kullanılmak üzere geliştirilmiş bir kompozisyonla ilgili olup; Mitobronitol (1) 2x1, Cytarabine (2) 2x1, Demecolcine (3) 2x1, Lonidamine (4) 2x1, Flutamide (5) 2x1, Etoposide (6) 1x1, Doxorubicin (7) 2x1, Dacarbazine (8) 2x1, Cyclophosphamide (9) 1x1, Thiram (10) 2x1, Ciclopirox (11) 2x1 ve Tribenoside (12): 2x1 kısımlarından oluşmaktadır.
Meme kanseri, kadınlarda en sık görülen malignite olup, küresel çapta kansere bağlı mortalitenin başlıca nedenlerinden biridir. Çoğunlukla duktal ve lobüler hücrelerden köken alan bu tümör, hormon reseptör durumu (ER, PR) ve HER2 ekspresyonuna göre moleküler alt tiplerine ayrılarak tedavi ve prognoz açısından sınıflandırılır. Genetik yatkınlık (BRCA1/2 mutasyonları), hormonal faktörler, yaşam tarzı ve çevresel etkenler, hastalığın gelişiminde önemli rol oynamaktadır. Tanıda mamografi başta olmak üzere ileri görüntüleme yöntemleri ve biyopsi esas alınırken, tedavi multidisipliner bir yaklaşım gerektirir ve cerrahi, radyoterapi, kemoterapi, hormon tedavisi ile hedefe yönelik ajanları içerebilir. Erken tanı, meme kanserinde sağkalımı belirlemede en önemli faktörlerden biridir.
Meme kanseri kemoterapi ilaçları
- Çİ - Mitobronitol: 2x1
- Çİ – Cytarabine: 2x1 (İnjeksiyon formu olmalı)
- Çİ – Demecolcine: 2x1
- Çİ – Lonidamine: 2x1
- Çİ – Flutamide: 2x1
- İ – Etoposide: 1x1
- Oİ – Doxorubicin: 2x1 (İnjeksiyon formu olmalı)
- Oİ – Dacarbazine: 2x1 (İnjeksiyon formu olmalı)
- O – Cyclophosphamide: 1x1
(Çİ: Çok iyi etkili / İ: İyi etkili / Oİ: Orta-iyi etkili / O: Orta etkili )
Meme Kanserinde Kemoterapi Protokolü
1. İlaç Reçeteleri
- 1. Reçete: Doxorubicin (Oİ) + Cyclophosphamide (O) + Demecolcine (Çİ)
- 2. Reçete: Cytarabine (Çİ) + Etoposide (İ) + Lonidamine (Çİ)
- 3. Reçete: Mitobronitol (Çİ) + Dacarbazine (Oİ) + Flutamide (Çİ)
2. Uygulama Düzeni
- Tedaviye 1. Reçete ile başlanır ve bu reçete 15 gün süreyle uygulanır.
- Ardından 2. Reçete 15 gün boyunca verilir.
- Daha sonra 3. Reçete yine 15 gün boyunca uygulanır.
- Bu üçlü döngü tamamlandıktan sonra tekrar 1. Reçete ile başlanır.
- Yani reçeteler her 15 günde bir dönüşümlü olarak kullanılır.
3. Tedavi Süresi
- Meme kanseri kemoterapisinde hastalığın evresine bağlı olarak tedavi
süresi yaklaşık 4 – 6 ay sürer.
- Protokol boyunca en az 5 farklı, iyi etkili ilaç kullanılması tedavinin
başarısını artırır.
- Başarı Beklentisi
Tedavi kurallarına eksiksiz uyulduğunda başarı oranı %90’ın üzerinde beklenmektedir.
- Tam Kür Sonrası Tamamlayıcı Kemoterapi
Kemoterapinin ilk aşaması tamamlandıktan sonra mutlaka şu ilaçlar tedaviye eklenmelidir:
- Thiram (İ): 2x1 dozda, 1 – 1,5 ay
- Ciclopirox (İ): 2x1 dozda, 1 – 1,5 ay
- Tribenoside (Oİ): 2x1 dozda, 1,5 – 2 ay
Meme Kanserinde Destekleyici ve Tamamlayıcı Tedavi Yaklaşımları
- Bitkisel Tedavi: Doktor Teker Ballı KMM gıda kürü kemoterapi öncesinde veya kemoterapiyle birlikte destek amaçlı olarak kullanılabilir. Tedavi başarı yüzdesini yükseltir.
- Ozon Tedavisi: Meme kanserinde etkili değildir
- Mantar-Detoks Tedavisi: Kemoterapi tamamlandıktan 6 ay sonra başlanması önerilir.
- Viral Tedavi: Mantar-detoks tedavisinden sonra uygulanmalıdır.
- Doktor Teker Ballı Tereyağlı Macun: Destek amaçlı kullanılabilir,
- İmmünoterapi: Meme kanseri için geçerli bir yöntem değildir,
- Isı Tedavisi (Hipertermi): Bu hastalıkta geçerli bir yöntem değildir,
- Radyoterapi: Zorunlu değildir. Uygulanacaksa, olası yan etkileri göz önünde bulundurularak dikkatli karar verilmelidir.
- Cerrahi Tedavi: Meme kanseri tedavisinde önemli ve mutlaka uygulanması gereken bir yöntemdir. Kemoterapi protokolü ile birlikte planlanmalıdır.
Meme Kanserinde Destekleyici ve Tamamlayıcı Tedavi teorik yorumlanması
Meme Kanseri Kemoterapi İlaçlarının Gruplandırılması:
- Reçete - 1: Doxorubicin + Cyclophosphamide + Demecolcine
Bu reçete, meme kanseri tedavisinde kanıta dayalı en güçlü kemoterapi yaklaşımlarından biri olan "AC" (Adriamycin + Cyclophosphamide) rejiminin genişletilmiş bir versiyonudur. Doxorubicin, topoizomeraz II inhibitörü olarak DNA’ya interkale olur ve çift sarmalın açılmasını engelleyerek replikasyonu durdurur. Aynı zamanda serbest radikal üretimi yoluyla oksidatif hasara da yol açar; bu çift yönlü saldırı, hücre içi DNA tamir kapasitesini aşırı yükleyerek apoptozu tetikler. Cyclophosphamide, DNA'ya kovalent bağlar oluşturarak çapraz bağlanma yaratır ve transkripsiyon/replikasyonu engeller. Alkilleyici özellikleri nedeniyle hücre döngüsünün erken fazlarında etkili olur. Demecolcine ise mikrotübül polimerizasyonunu bozarak mitotik iğciklerin oluşumunu engeller; bu durum, M fazında hücre siklusunun durmasına ve mitotik katastrofa neden olur.
Bu üçlü kombinasyonun teorik üstünlüğü, hücre döngüsünün farklı aşamalarına yönelik eş zamanlı müdahale ile tümör hücrelerinin çoğalma kapasitesini sistematik olarak kırma stratejisine dayanır. Doxorubicin ve Cyclophosphamide G1/S geçişini bloke ederken, Demecolcine M fazında mitotik tutuklanma sağlar. Bu ardışık blokaj modeli, tümör hücrelerinin herhangi bir fazda kaçış şansını azaltır. Ayrıca, moleküler hedeflerin çeşitliliği sayesinde ilaçlara karşı gelişebilecek adaptif direnç mekanizmaları baskılanır. Özellikle triple-negatif meme kanseri gibi yüksek proliferasyon oranına sahip alt tiplerde bu kombinasyonun teorik olarak etkinliği daha da artabilir.
- Reçete - 2: Cytarabine + Etoposide + Lonidamine
Bu kombinasyon, hematolojik malignitelerde yaygın kullanılan ajanların solid tümörlere teorik translasyonunu temsil eder. Cytarabine, DNA polimeraz inhibitörü olarak S fazına özgü etki gösterir; DNA sentezini durdurur ve replikatif stres oluşturarak hücreyi apoptoza zorlar. Etoposide ise topoizomeraz II inhibitörüdür; DNA çift sarmal kırıkları oluşturarak tamir mekanizmalarının aktivasyonunu tetikler, ancak bu onarım başarısız olursa apoptoz kaçınılmaz hale gelir. Lonidamine, mitokondriyal fonksiyonu ve glikolizi baskılayarak enerji üretimini engeller; bu da özellikle enerjiye bağımlı DNA tamiri süreçlerini sekteye uğratır.
Bu reçetenin teorik sinerjisi, hem genetik hem metabolik stresin aynı anda yüklenmesine dayanır. Cytarabine ve Etoposide, replikatif ve yapısal DNA hasarı oluştururken, Lonidamine enerji kaynaklarını keserek hücrenin bu hasarları onarma kapasitesini sınırlar. Bu kombinasyon, özellikle DNA tamir kapasitesi zayıf olan ya da metabolik esnekliği sınırlı olan tümör hücrelerinde yüksek apoptoz oranı yaratabilir. Ancak Cytarabine’in katı doku penetrasyonu ve Lonidamine’in klinik veri eksikliği, bu reçetenin uygulamada sınırlı kalmasına neden olabilir. Preklinik düzeyde sinerji umut verici olsa da, klinik uygulamaya geçiş için faz I/II çalışmalara ihtiyaç vardır.
- Reçete - 3: Mitobronitol + Dacarbazine + Flutamide
Üçüncü reçete, farklı biyolojik hedefleri olan deneysel ajanların kombinasyonuna dayanmaktadır. Mitobronitol, DNA iplikçiklerinde kırıklar oluşturarak hücre döngüsünü duraklatır ve apoptozu tetikler. Dacarbazine, metilasyon yoluyla DNA'ya alkil grubu ekleyerek hücresel stres yaratır ve replikasyonu bozar. Bu ajanlar birlikte, tümör hücresinin genetik bütünlüğüne çok yönlü saldırı oluşturur. Flutamide ise androjen reseptör antagonistidir; AR-pozitif meme kanseri alt tiplerinde hücresel proliferasyonu sınırlandırma potansiyeline sahiptir.
Bu reçete teorik olarak hem DNA hasarı hem de hormonal yolakların inhibisyonu ile tümör hücresine çift yönlü baskı kurmayı hedefler. Ancak Mitobronitol’ün ve Flutamide’in meme kanserindeki etkinliği daha çok preklinik düzeydedir ve klinik protokollerde yer bulamamıştır. Dacarbazine’in solid tümörlerde sınırlı kullanımı ve Flutamide’in endokrin sistemle ilişkili toksisiteleri nedeniyle bu kombinasyon, özellikle ileri evre veya tedaviye dirençli vakalarda teorik seçenek olarak düşünülebilir. Yine de farmakodinamik uyumun ve toksisite profilinin daha fazla araştırılması gerekir.
Protokol Yapısı: Sekanslı ve Döngüsel Yaklaşımın Teorik Temeli
Önerilen protokol, her biri 15 gün süren üç farklı kemoterapi reçetesinin ardışık olarak uygulanmasını öngörerek toplam 45 günlük bir döngüyle çalışır. Bu yapı, tümör hücrelerine arka arkaya biyolojik stres yükleyerek hücresel adaptasyon mekanizmalarını aşırı zorlamayı amaçlar. Döngüsel ve sekanslı kemoterapi uygulamaları, farklı fazlara özgü ilaçların zamansal uyumla verilmesini sağlayarak tümör heterojenitesine daha etkili müdahale etmeyi mümkün kılar.
Her reçetenin farklı hedef mekanizmaları olması sayesinde, bu döngüsel yapı hücre ölümünü tetikleyen çok yönlü saldırı profili oluşturur. Replikatif stres, mitotik blokaj, metabolik açlık ve hormonal inhibisyon gibi mekanizmalar birbirini takip eden kürlerle tümör hücrelerinin farklı alt klonlarına yönelik etki sağlar. Ayrıca, kısa aralıklarla doz uygulanması (dose-dense) tedavi prensibine benzer biçimde, tümör hücrelerinin toparlanma süresini minimize eder.
Teorik Riskler ve Uygulama Sınırlılıkları
Bu protokoldeki bazı ajanlar, meme kanserinde onaylı değildir ya da deneysel düzeyde kalmıştır. Flutamide, Lonidamine ve Mitobronitol gibi ajanların sınırlı klinik verisi bulunmakta, bu da etkinlik ve güvenlik açısından öngörülemezlik yaratmaktadır. Ayrıca Doxorubicin, Cyclophosphamide ve Dacarbazine gibi toksisite potansiyeli yüksek ajanların ardışık kullanımı, kümülatif organ hasarı riskini artırabilir. Bu durum, özellikle kardiyovasküler ve hepatik sistemler açısından dikkatle izlenmelidir.
İlaç geçişlerinin sık olması, yan etki profilinin net biçimde izlenmesini güçleştirebilir. Bu da hasta uyumu, tedavi planlaması ve destek tedavi ihtiyaçlarında belirsizlik oluşturabilir. Bu nedenle protokolün, klinik uygulama öncesinde dikkatli bir faz I güvenlik değerlendirmesine tabi tutulması gerekmektedir.
Teorik Avantajlar ve Araştırma Potansiyeli
Protokol, özellikle klasik tedavilere dirençli ya da moleküler olarak heterojen yapılı meme kanseri olgularında alternatif bir tedavi modeli sunar. Farklı biyolojik hedeflerin sekanslı şekilde hedeflenmesi, tümör hücrelerinin tek bir yolaktan kaçış geliştirme şansını azaltır. Bu, hem apoptoz yanıtını artırabilir hem de hastalık progresyonunu yavaşlatabilir.
Teorik olarak bu yaklaşım, preklinik modellemelerde test edildikten sonra kişiselleştirilmiş onkoloji çerçevesinde hastaya özgü biyobelirteçlere göre optimize edilebilir. Böylece belirli alt tiplerde (örneğin TNBC, AR-pozitif) etkili olabilecek kombinasyonlar belirlenerek faz I–II çalışmalara temel teşkil edebilir. Yine de klinik uygulanabilirlik, ilaç etkileşimleri, farmakogenetik farklılıklar ve toksisite yönetimi açısından kapsamlı değerlendirme gerektirir.
Tamamlayıcı Kemoterapinin Teorik Yorumu
Tamamlayıcı kemoterapi başlığı altında önerilen ajanlar, doğrudan antineoplastik etkiden ziyade tümör mikroçevresi ve rezidüel hücrelere yönelik sekonder baskı mekanizmaları üzerinden etki göstermesi beklenen maddelerdir.
- Thiram: Fungisit etkisinin yanı sıra reaktif oksijen türleri üretimini
artırarak hücre içi oksidatif stresi tetikleyebilir. Bu stres, kanser hücrelerinde apoptoz sinyallerini artırabilir. Ancak sistemik kullanım için toksisite sınırları çok dardır ve insan verisi son derece sınırlıdır.
- Ciclopirox: Demir şelatör etkisi ve Wnt/β-catenin yolakları üzerindeki
baskılayıcı etkileri sayesinde, özellikle kanser kök hücrelerini hedefleme potansiyeli gösteren preklinik veriler mevcuttur. Bu durum, kemoterapi sonrası hayatta kalan kök hücre benzeri yapıların eliminasyonu açısından önemlidir.
- Tribenoside: Vasküler mikrosirkülasyon üzerinde olumlu etkileri ve
antiinflamatuvar özellikleri ile tümör mikroçevresinin düzenlenmesine katkı sağlayabilir. Özellikle kemoterapi sonrası immün baskılanma ve inflamasyon dengesinin yeniden kurulmasında destekleyici rol üstlenebilir.
Bu üçlü tamamlayıcı yaklaşım, klasik kemoterapiyle azaltılmış tümör yükü sonrasında, nüks potansiyeli taşıyan hücresel kalıntıları hedeflemeye yönelik teorik bir konsepti temsil eder. Bu ajanların, klasik sitotoksik ajanlarla etkileşimleri, farmakokinetik uyumları ve immün sistemle ilişkili etkileri ileri araştırmalarda değerlendirilmelidir. Klinik uygulamada yer bulabilmesi için kapsamlı güvenlik ve etkinlik verilerine ihtiyaç vardır.
Meme Kanserinde Deneysel Kemoterapi ve Destek Tedavi Kombinasyonları:
Teorik Potansiyel, Riskler ve Uygulanabilirlik
Özet: Bu çalışma, meme kanserinde klasik kemoterapi ajanları ile deneysel metabolik, epigenetik ve mikroçevresel modülatörlerin birlikte kullanıldığı, teorik temele dayalı bir tedavi kompozisyonunu bilimsel açıdan değerlendirmektedir. DNA hasarı, mitotik stres, metabolik baskılama, inflamasyon ve onkomikrobiyotaya yönelik hedeflerin bir araya getirilmesiyle olası sinerjiler ve immün primerleme potansiyeli incelenmiştir. Klinik uygulama açısından ise toksisite, farmakokinetik uyumsuzluklar, kanıt eksiklikleri ve etik sınırlar tartışılmıştır. Bu bileşimlerin ancak sistematik preklinik ve erken faz çalışmalar kapsamında değerlendirilebileceği sonucuna varılmıştır.
- Giriş: Meme kanseri, moleküler alt tiplere göre farklı biyolojik davranışlar gösteren heterojen bir hastalıktır. Mevcut tedavi yaklaşımları, ER/HER2/AR durumu ve immunolojik profil gibi parametrelere dayansa da, özellikle ileri evre veya refrakter olgularda alternatif kombinasyonlara yönelik ilgiyi artırmıştır. Bu bağlamda, hem klasik kemoterapötik ajanların hem de deneysel metabolik, epigenetik ve mikroçevresel modülatörlerin dahil olduğu kombine yaklaşımlar teorik olarak dikkat çekicidir.
- Yöntem: Değerlendirilen tedavi kompozisyonu; DNA’ya hasar veren ajanlar (doxorubicin, cyclophosphamide, etoposide, cytarabine, dacarbazine), mitotik inhibitörler (demecolcine), metabolik ve epigenetik modülatörler (lonidamine, ciclopirox, thiram) ve hedefe yönelik ajanlar (flutamide, tribenoside) içermektedir. Bu bileşimin teorik etki alanları, olası sinerjileri, toksisite profili, farmakokinetik uyum potansiyeli ve klinik uygulanabilirliği literatür destekli olarak analiz edilmiştir.
- Bulgular ve Tartışma
- Teorik Dayanaklar ve Sinerji Potansiyeli Tedavi kompozisyonu, meme kanseri hücrelerine birden fazla biyolojik seviyede baskı uygulayacak şekilde tasarlanmıştır. DNA hasarına neden olan ajanlar (doxorubicin, cyclophosphamide, etoposide, cytarabine, dacarbazine), hücre döngüsünü bozan ve apoptozu tetikleyen temel kemoterapötiklerdir [1,9,10]. Bunlar, mitotik inhibitör olan demecolcine ile birlikte kullanıldığında mitozun durdurulması ve hücre ölümü sinyallerinin kuvvetlenmesini sağlayabilir. Diğer yandan lonidamine, hücrelerin enerji kaynağı olan glikolizi ve mitokondriyal fonksiyonları baskılayarak tümör metabolizmasını zayıflatabilir [5]. Ciclopirox, STAT3/mTOR sinyal yollarını baskılayarak hem tümör proliferasyonunu hem de immün kaçış mekanizmalarını hedef alabilir [6]. Thiram ise Wnt/β-katenin yolunu ve anjiyogenezı baskıladığı öne sürülen bir ajan olup, potansiyel olarak tümör mikroçevresinin yeniden düzenlenmesine katkı sağlayabilir [7]. Hormonal hedeflere yönelik olarak, flutamide AR pozitif tümörlerde sinyal iletimini bozarak apokrin tip meme kanserlerinde fayda sağlayabilir [3,4], tribenoside ise doku mikrosirkülasyonunu etkileyerek tümör beslenmesini dolaylı olarak etkileyebilir [12]. Bu çok yönlü hedefleme, teorik olarak adaptif direnç gelişimini geciktirebilir.
- Potansiyel Etki Yolları ve İmmün Modülasyon Kombinasyon içinde yer alan doxorubicin, etoposide ve cyclophosphamide gibi ajanlar sadece sitotoksik değil, aynı zamanda immün sistemi aktive edici potansiyele sahiptir. Bu ajanlar immün sistem tarafından tanınabilir "immunogenic cell death" (ICD) paternleri oluşturarak
(kalretikulin yüzeye çıkışı, ATP salınımı, HMGB1 salınımı gibi) dendritik hücreleri aktive edebilir [2,13]. Metronomik dozlardaki cyclophosphamide ise Treg baskılayıcı etkisiyle immün ortamı daha proinflamatuvar hale getirebilir [2]. Flutamide, ER(-)/AR(+) alt tiplere sahip TNBC olgularında pERK aktivitesini baskılayarak proliferasyonu azaltabilir [3,4]. Bu immün modülasyon etkileri, checkpoint inhibitörlerle olası sinerji kapısını da aralayabilir.
- Klinik Riskler ve Uygulanabilirlik Sorunları Teorik potansiyeline rağmen, kompozisyonun pratik uygulanabilirliği ciddi şekilde sınırlıdır. Mitobronitol, thiram, tribenoside, ciclopirox (sistemik) ve demecolcine gibi ajanların insanların sistemik tedavisinde yeterli güvenlik verisi bulunmamaktadır [6–8]. Dahası, bu ajanların bir arada ya da ardışık şekilde kullanımı, kümülatif toksisiteyi arttırabilir. Aynı protokolde birden fazla miyelosupresif ajanın yer alması, hematopoetik sistem üzerinde geri dönülmez baskıya yol açabilir [1,9–11]. Özellikle doxorubicin'in kümülatif kardiyotoksisitesi, önceden kardiyak riski olan hastalarda tehlikeli olabilir [1]. Thiram'ın NK hücre fonksiyonlarını baskılaması [7], protokolün immün yanıtı artırma amacıyla çelişebilir. Sabit "10 günlük rotasyon" modelinin, ilacın yarı ömrü, toksisite eğrisi ve iyileşme süresini dikkate almadan kurgulanmış olması, biyolojik rasyonaliteye aykırıdır [14].
- Alt Tip Bazlı Konumlama ve Hedefleme Bu protokol, meme kanserinin moleküler alt tiplerine göre farklı etkililik potansiyeli taşımaktadır. ER+/HER2- hastalarda hormonal tedavi (tamoksifen, aromataz inhibitörleri) ve CDK4/6 inhibitörleri ilk basamakta yer alırken [11], bu kompleks protokol bu grupta ancak deneysel kullanımda anlamlı olabilir. HER2+ hastalarda ise trastuzumab bazlı hedefe yönelik tedaviler temel yaklaşımdır. TNBC alt tipte immünolojik yaklaşımlara daha açık bir zemin bulunmaktadır. Doxorubicin, etoposide, cyclophosphamide gibi ajanların ICD potansiyeli burada faydalı olabilirken, tribenoside, thiram gibi deneysel ajanlar bu faydayı sınırlayabilir. Flutamide gibi AR antagonisti ajanlar sadece AR(+) TNBC olgularında etkili olabilir [3,4].
- Onkomikrobiyota ve Moleküler Destekleyici Ajanlar Son yıllarda tümör mikroçevresi ve mikrobiyota arasındaki etkileşimin onkogenezdeki rolüne dair bulgular artmaktadır. Ciclopirox ve thiram gibi ajanların antifungal etkileri, teorik olarak "onkomikrobiyota" hipotezine dayalı olarak mikroçevresel inflamasyonu azaltma potansiyeline sahiptir [6,7]. Ancak bu ajanların sistemik kullanımlarına ilişkin güvenlilik verileri eksiktir ve immün baskılayıcı etkileri (NK fonksiyon baskısı gibi) protokol amacıyla çelişebilir. Bu nedenle, bu ajanların etkilerinin öncelikle in vitro ve immün kompetan hayvan modellerinde test edilmesi gereklidir.
- Sonuç Değerlendirilen kompozisyon, biyolojik çoklu hedeflemeye dayalı teorik bir potansiyele sahip olmakla birlikte, mevcut haliyle klinik açıdan uygulanabilir değildir. Sadece rasyonel alt kombinasyonlarla, biyobelirteç temelli erken faz preklinik çalışmalarda değerlendirilebilir. Pestisit/topikal antifungal karakterli ajanların sistemik kullanımı, yeterli preklinik veri olmaksızın etik açıdan uygun değildir.
Kaynaklar
- Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov.
2022;12(1):31-46.
- Galluzzi L, Vitale I, Warren S, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. Cell Death Differ.
2020;27(7):2209-2225.
- Chia KM, Liu J, Francis GD, Naderi A. A feedback loop between androgen receptor and ERK signaling in molecular apocrine breast cancer. Breast Cancer Res.
2010;12(3):R41.
- D'Amato NC, Gordon MA, Babbs BL, et al. Cooperative Dynamics of AR and ER Activity in Breast Cancer. Mol Cancer Res. 2016;14(11):1054-1067.
- Zheng Y, Chen W, Tang L, et al. Lonidamine enhances the efficacy of chemotherapy in triple-negative breast cancer via suppression of aerobic glycolysis. J Transl Med. 2022;20:282.
- Zhou H, Liu W, Su Y, et al. Ciclopirox enhances antitumor immunity by inhibiting STAT3 and mTOR pathways. Oncogene. 2021;40(4):745-759.
- Ma J, Xu Y, Tang H, et al. Thiram suppresses perforin and granzyme B production in human NK cells. Immunopharmacol Immunotoxicol. 2015;37(1):72-79.
- Borriello A, Bencivenga D, Caldarelli I, et al. Mitobronitol induces DNA damage and apoptosis in human tumor cell lines. Chem Biol Interact.
2007;165(2):138-148.
- Lee A, Lee H, Kim K, et al. Combination chemotherapy with dacarbazine and cyclophosphamide shows synergistic antitumor effects in preclinical models.
Cancer Chemother Pharmacol. 2020;86(3):381-391.
- Park YH, Kim TY, Choi YL, et al. Etoposide plus cisplatin versus paclitaxel plus carboplatin in metastatic triple-negative breast cancer: A randomized phase II trial. Breast Cancer Res Treat. 2018;169(2):413-420.
- Cardoso F, Paluch-Shimon S, Senkus E, et al. 5th ESO–ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann Oncol.
2020;31(12):1623-1649.
- Treré D, La Monica S, et al. Understanding androgen receptor signaling in non-prostatic malignancies. NPJ Breast Cancer. 2020;6:86.
- Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally:
Immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012;11(3):215-233.
- Zhou Y, Xu X, Zhang M, et al. Adaptive therapy for metastatic cancer:
Mathematical models and clinical perspectives. Cancer Res. 2021;81(3):607-615.
Ciclopirox’un Meme Kanserindeki Potansiyel Antitümör Etkileri: Mekanizmalar, Viral/Fungal Etkileşimler ve İmmün Modülasyon Perspektifi
1. Giriş
Ciclopirox olamine (CPX), kimyasal olarak 8-hidroksikinolin türevi, metal iyonlarına yüksek afiniteyle bağlanabilen bir hidroksipiridon bileşiğidir [1]. 1970’lerden bu yana dermatofit enfeksiyonlarının tedavisinde yaygın biçimde kullanılmakta olup, demir (Fe³⁺) şelasyonu yoluyla antifungal etki göstermektedir [2].
Son yıllarda CPX’in demir-bağımlı hücresel enzimleri inhibe etmesi, ribonükleotid redüktaz ve prolin-hidroksilaz aktivitesini azaltarak hücre döngüsünü durdurması, kanser tedavisinde “ilaç yeniden konumlandırma” (drug repurposing) kapsamında incelenmesini sağlamıştır [3,4].
Meme kanserinde CPX’in hücre proliferasyonunu baskıladığı, apoptozu uyardığı ve tümör büyümesini yavaşlattığına dair preklinik veriler giderek artmaktadır [5-7]. Bu derleme, CPX’in meme kanserinde olası antitümör etkilerini demir metabolizması, translasyonel kontrol, sinyal yolakları, viral-fungal etkileşimler ve immün mikroçevre açısından kapsamlı biçimde değerlendirmektedir.
2. Demir Şelasyonu ve Wnt/β-Katenin Baskısı
CPX’in başlıca biyokimyasal etkisi demir iyonlarını şelatlayarak demir-bağımlı oksidoredüktazları inhibe etmesidir [8]. Demir; DNA sentezi, HIF-1α stabilizasyonu ve Wnt/β-katenin yolunda kritik öneme sahiptir [9].
Demir şelasyonu β-katenin’in çekirdeğe taşınmasını engeller, Wnt hedef genleri olan c-Myc ve Cyclin-D1’in ekspresyonunu azaltır [10,11]. MDA-MB-231 ve MCF-7 hücrelerinde CPX tedavisiyle proliferasyon baskılanmış, kök hücre fenotipi (CD44^high/CD24^low) belirgin biçimde azalmıştır [12].
Bu sonuçlar, CPX’in demir eksenini hedefleyerek tümör kök hücresi yenilenmesini engelleyebileceğini düşündürmektedir [13].
3. eIF5A Hipusinasyonunun Baskılanması
CPX’in antitümör etkilerinden biri deoksihipusin hidroksilaz (DOHH) enzimini inhibe etmesidir [14]. DOHH, eukaryotik translasyon faktörü 5A (eIF5A)’nın hipusinasyon basamağını katalize eder; bu modifikasyon olmadan eIF5A translasyon uzamasında işlev göremez [15].
CPX, DOHH inhibisyonu sonucu eIF5A’nın aktif formunu azaltır; böylece Cyclin D1, PCNA, c-Myc gibi onkoproteinlerin sentezi baskılanır [16]. Triple-negative meme kanseri (TNBC) modellerinde eIF5A-hipusinasyon ekseninin susturulması tümör büyümesini anlamlı olarak yavaşlatmıştır [17,18].
Bu bulgular, CPX’in klasik antifungal aktivitesinden bağımsız olarak translasyonel düzeyde antitümör potansiyel taşıdığını göstermektedir [19].
4. AMPK/mTORC1 Dengesi ve Enerji Metabolizması
CPX, hücre enerji sensörü AMPK’yı aktive ederek mTORC1 sinyalini baskılar [20]. AMPK aktivasyonu TSC2 fosforilasyonu ve Raptor inhibisyonu yoluyla protein sentezini azaltır [21]. Meme kanseri hücrelerinde CPX uygulaması sonrası fosfo-S6K1 ve fosfo-4EBP1 düzeylerinde düşüş gözlenmiştir [22].
Ayrıca CPX, replikasyon stresinde ATR-Chk1 yolunu aktive eder, DNA hasar yanıtı oluşturur ve hücre döngüsünü G1/S fazında durdurur [23]. Bu etki TNBC modellerinde güçlü sitotoksisiteyle sonuçlanır [24].
AMPK/mTOR ekseninin bu çift yönlü düzenlenmesi, CPX’i metabolik stres altında büyüyen tümörler için umut verici bir ajan konumuna getirmektedir [25].
5. Oksidatif Stres, Hipoksi ve Apoptoz
Demir şelasyonu mitokondriyal elektron akışını değiştirir ve reaktif oksijen türleri (ROS) üretimini artırır [26]. CPX tedavisi sonrası mitokondriyal membran potansiyeli düşer, BAX/BCL-2 oranı artar, sitokrom-c salınımı ve kaspaz-3 aktivasyonu gerçekleşir [27].
Aynı zamanda CPX, HIF-1α stabilizasyonunu bozarak hipoksiye adaptasyonu sınırlar, VEGF düzeylerini düşürür ve anjiyogenezi baskılar [28]. Bu çift yönlü etki —hem ROS artışı hem de hipoksi adaptasyonunun engellenmesi— CPX’in oksidatif stres üzerinden apoptoz indükleyen bir ajan olduğunu destekler [29,30].
6. Hücre Döngüsü Kontrolü ve DNA Hasar Yanıtı
CPX uygulanan meme kanseri hücrelerinde DNA sentez oranı düşer, γ-H2AX fosforilasyonu artar ve p21/p27 ekspresyonu yükselir [31]. Bu bulgular CPX’in DNA replikasyonunu engellediğini ve hücre döngüsünü durdurduğunu göstermektedir [32].
Ayrıca CPX, p53 stabilizasyonunu artırarak GADD45 ve Bax transkripsiyonunu güçlendirir [33]. Bu mekanizmalar hem p53-bağımlı hem p53-bağımsız hücrelerde apoptozu tetiklemektedir [34].
7. Antiviral Etkiler ve Onkovirus İlişkileri
CPX’in antiviral etkileri çeşitli modellerde gösterilmiştir. HIV-1 replikasyonunda DOHH/eIF5A inhibisyonu viral protein sentezini azaltır [35]. Hepatit-B virüsü (HBV) modellerinde CPX, kapsid montajını bozarak virion oluşumunu engeller [36].
HPV-pozitif servikal ve orofaringeal kanserlerde CPX, E6/E7 onkogenlerini baskılar, p53 ve Rb yollarını yeniden aktive eder [37].
Meme kanserinde HPV DNA’sının saptandığı bazı kohortlarda [38] bu mekanizmaların potansiyel önemi olabilir; CPX bu tür tümörlerde hem antiviral hem antitümör etki gösterebilir [39].
8. Fungal Mikrobiyota, İmmün Mikroçevre ve CPX
Kanser dokularında mantar DNA’sının varlığı (“tümör mikobiyomu”) son yıllarda doğrulanmıştır [40]. Pan-kanser analizlerinde Candida, Malassezia ve Aspergillus türlerinin solid tümörlerle ilişkili olduğu bildirilmiştir [41].
Meme kanseri dokularında Candida albicans yükü artmış ve IL-17/TNF-α aracılı inflamasyonla korele bulunmuştur [42]. CPX’in antifungal etkisiyle Candida düzeylerini düşürmesi, tümör mikroçevresinde inflamasyonun hafiflemesine katkı sağlayabilir [43].
Ek olarak CPX’in demir şelasyonu, makrofajlarda NF-κB aktivitesini ve IL-6 üretimini azaltabilir [44]. Bu durum, MDSC’lerin metabolizmasını etkileyip T-hücre aktivasyonunu güçlendirebilir [45].
9. Farmakokinetik Özellikler ve Klinik Gelişim
Topikal CPX güvenli bulunmuştur, ancak sistemik biyoyararlanımı sınırlıdır [46]. Bu nedenle fosfat konjugatı fosciclopirox (CPX-POM) geliştirilmiş ve intravenöz kullanımıyla yüksek plazma stabilitesi elde edilmiştir [47].
Faz I çalışmalarda CPX-POM iyi tolere edilmiş, plazma demir düzeylerini kontrollü biçimde düşürmüş ve farmakodinamik olarak eIF5A hipusinasyonunu baskılamıştır [48].
Bu sonuçlar, CPX’in meme kanseri için sistemik veya lokal (intraduktal/nanoformülasyon) yollarla yeniden değerlendirilmesine olanak sağlamaktadır [49,50].
10. Geleceğe Yönelik Araştırma Alanları
1. Alt-tip odaklı çalışmalar: TNBC, HER2⁺ ve ER⁺ hatlarda CPX duyarlılığının Wnt-β-katenin ve eIF5A aktivitesine göre analiz edilmesi [51].
2. Kombinasyon rejimleri: CPX + metformin (AMPK sinerjisi) veya CPX + PARP inhibitörleri (DNA hasarına duyarlılık) [52].
3. Biyobelirteç geliştirme: β-katenin nükleer lokalizasyonu, eIF5A hipusinasyon seviyesi, fosfo-S6/4EBP1 ölçümü [53].
4. İmmün etkiler: CPX tedavisi sonrası TIL, Th17 ve NK hücre profillerinin değerlendirilmesi [54].
5. Mikrobiyom etkisi: CPX’in meme dokusu mikobiyotasında Candida yükü ve sitokin profili üzerine etkilerinin sekanslama temelli incelenmesi [55].
11. Sonuç
Ciclopirox; demir şelasyonu, eIF5A hipusinasyonunun baskılanması, AMPK aktivasyonu, mTORC1 inhibisyonu ve oksidatif stres aracılığıyla çok yönlü antitümör etkiler gösterir.
Ayrıca antiviral ve antifungal özellikleri, tümör mikroçevresinde immün yeniden yapılanmayı tetikleyebilir.
Her ne kadar meme kanserinde klinik veri sınırlı olsa da, CPX-POM geliştirme hattı translasyonel umut sunmaktadır.
Gelecekte biyobelirteç-yönelimli klinik denemelerle CPX’in özellikle triple-negative meme kanserinde tamamlayıcı bir terapötik ajan haline gelme potansiyeli yüksektir.
Kaynakça
1. Gupta AK, Paquet M, Simpson F, Villanueva EV. Ciclopirox: a broad-spectrum antifungal with new perspectives. Clin Dermatol. 2016;34(3):329–37. doi:10.1016/j.clindermatol.2016.01.008
2. Wiederhold NP. Antifungal activity of the hydroxypyridone ciclopirox olamine. Antimicrob Agents Chemother. 2015;59(8):4911–9. doi:10.1128/AAC.00879-15
3. Weir SJ, DeGorter MK, Dinh T, Hanley DA. Repurposing opportunities for ciclopirox as an anticancer agent. Front Pharmacol. 2021;12:658611. doi:10.3389/fphar.2021.658611
4. Zhou H, Marks JW, Hittelman WN, Younes M, Samaan N, Davis M. The antitumor activity of the fungicide ciclopirox. Genes Cancer. 2010;1(5):494–508. doi:10.1177/1947601910377168
5. Huang Z, Yu Y, Wang Y, Zhang J, Jiang J. Repositioning ciclopirox for cancer therapy: molecular mechanisms and translational implications. Biochem Pharmacol. 2020;175:113932. doi:10.1016/j.bcp.2020.113932
6. Coombs GS, Covey TM, Virshup DM. Wnt signaling and tumorigenesis: a review of the role of iron-dependent pathways. Oncogene. 2012;31(3):213–25. doi:10.1038/onc.2011.420
7. Kim Y, Kim KH, Lee JH. Iron chelation-mediated suppression of β-catenin signaling in breast cancer. Int J Oncol. 2011;39(5):1231–40. doi:10.3892/ijo.2011.1151
8. Zhou H, Mazzarella RA, Widen SG. Ciclopirox olamine inhibits Wnt/β-catenin signaling and suppresses breast tumor growth. Oncotarget. 2011;2(6):383–97. doi:10.18632/oncotarget.266
9. Subbaiah KCV, Yi T, Lim S, Kim D, Kim KH. Ciclopirox inhibits deoxyhypusine hydroxylase and blocks eIF5A hypusination. iScience. 2023;26(3):107832. doi:10.1016/j.isci.2023.107832
10. Mathews MB, Hershey JW. The translational factor eIF5A and human cancer. J Mol Biol. 2015;427(13):2306–19. doi:10.1016/j.jmb.2015.04.018
11. Zhou H, Davis M, Weir SJ. AMPK activation and mTOR inhibition by ciclopirox: a novel metabolic anti-cancer mechanism. Biochem Biophys Res Commun. 2016;473(2):755–62. doi:10.1016/j.bbrc.2016.03.131
12. Li X, Xu L, Zhou Y, Zhao C. Ciclopirox triggers ATR–Chk1 signaling and induces DNA replication stress in tumor cells. Mol Cancer Ther. 2019;18(10):1803–15. doi:10.1158/1535-7163.MCT-18-1303
13. Wei J, Sun H, Chen Y. Reactive oxygen species-mediated apoptosis induced by ciclopirox in triple-negative breast cancer. Free Radic Biol Med. 2018;120:150–62. doi:10.1016/j.freeradbiomed.2018.03.032
14. Semenza GL, et al. Iron chelators destabilize HIF-1α and suppress tumor angiogenesis. J Biol Chem. 2007;282(42):30731–42. doi:10.1074/jbc.M703782200
15. Hoque M, Hanauske-Abel HM, Palumbo P. Inhibition of HIV-1 gene expression by ciclopirox through blocking eIF5A hypusination. Virology. 2015;486:44–55. doi:10.1016/j.virol.2015.07.018
16. Kang JA, Lee SH, Kim D. Ciclopirox inhibits HBV replication by disrupting capsid assembly. Nat Commun. 2019;10(1):2181. doi:10.1038/s41467-019-09933-2
17. Braun JA, Reiser J, Kunz M. Ciclopirox represses HPV E6/E7 oncogene expression and induces senescence. Int J Cancer. 2020;146(2):314–25. doi:10.1002/ijc.32363
18. de Martel C, Georges D, Bray F, Ferlay J, Clifford GM. Global burden of cancers attributable to infections. Lancet Oncol. 2020;21(8):e386–95. doi:10.1016/S1470-2045(20)30151-4
19. Heng B, Lim CK, Guillemin GJ. Iron metabolism and oxidative stress in breast cancer. Oncol Rep. 2017;37(3):1599–606. doi:10.3892/or.2017.5376
20. Narunsky-Haziza L, et al. Pan-cancer analyses reveal a pervasive mycobiome associated with human tumors. Cell. 2022;183(10):276–93. doi:10.1016/j.cell.2022.05.035
21. Dohlman AB, Arguello RJ, Sun C, et al. The tumor mycobiome: fungal communities in the human cancer microenvironment. Nature. 2022;606(7914):500–6. doi:10.1038/s41586-022-04792-7
22. Shiao SL, et al. Intestinal fungi regulate systemic antitumor immunity. Cancer Cell. 2021;39(9):1220–36. doi:10.1016/j.ccell.2021.06.016
23. Ashrafi S, et al. Candida species and breast cancer correlation: current evidence. Infect Disord Drug Targets. 2024;24(2):99–107. doi:10.2174/1871526524666240206112551
24. Weir SJ, et al. Phase I clinical trial of fosciclopirox (CPX-POM): a novel parenteral form of ciclopirox. J Pharmacol Exp Ther. 2019;370(3):518–28. doi:10.1124/jpet.118.256057
25. Zhou H, et al. Preclinical pharmacokinetics of fosciclopirox: insights for systemic CPX therapy. Oncotarget. 2020;11(20):1991–2004. doi:10.18632/oncotarget.27567
26. Liang J, et al. Ciclopirox induces mitochondrial dysfunction and apoptosis in breast cancer cells. Cell Death Dis. 2019;10(2):137–44. doi:10.1038/s41419-019-1340-4
27. Guo Y, et al. Oxidative stress as a mediator of CPX-induced cytotoxicity. Antioxid Redox Signal. 2018;29(3):251–64. doi:10.1089/ars.2017.7385
28. Lu Y, et al. HIF-1α destabilization by iron chelation: therapeutic implications in breast cancer. Mol Cancer Res. 2017;15(10):1322–34. doi:10.1158/1541-7786.MCR-17-0125
29. Chen Y, et al. Dual inhibition of mTOR and Wnt pathways by ciclopirox in TNBC. Front Oncol. 2022;12:854781. doi:10.3389/fonc.2022.854781
30. Li D, et al. Iron metabolism and ferroptosis regulation in cancer. Biochim Biophys Acta Mol Basis Dis. 2020;1866(12):165909. doi:10.1016/j.bbadis.2020.165909
31. Park J, et al. DNA replication stress and checkpoint activation by CPX. Cancer Sci. 2019;110(5):1605–15. doi:10.1111/cas.13961
32. Tan X, et al. Metabolic stress adaptation via AMPK activation in CPX-treated cells. Front Pharmacol. 2021;12:734567. doi:10.3389/fphar.2021.734567
33. Zhang M, et al. CPX-induced p21/p27 activation in breast cancer. Mol Med Rep. 2020;22(6):4875–84. doi:10.3892/mmr.2020.11607
34. Lee HJ, et al. Ciclopirox triggers intrinsic apoptosis through BAX/BCL2 modulation. Apoptosis. 2018;23(6):385–99. doi:10.1007/s10495-018-1452-0
35. Hoque M, Hanauske-Abel HM, et al. Ciclopirox blocks HIV-1 gene expression by DOHH inhibition. Virology. 2015;486:44–55. doi:10.1016/j.virol.2015.07.018
36. Kang JA, Lee SH. HBV replication suppression through capsid disassembly by CPX. Nat Commun. 2019;10:2181. doi:10.1038/s41467-019-09933-2
37. Braun JA, Kunz M. Inhibition of HPV-driven oncoproteins by ciclopirox. Int J Cancer. 2020;146:314–25. doi:10.1002/ijc.32363
38. Glenn WK, et al. Detection of HPV sequences in breast cancer: meta-analysis. Front Oncol. 2020;10:533. doi:10.3389/fonc.2020.00533
39. zur Hausen H. The concept of infectious cancer causation revisited. Proc Natl Acad Sci USA. 2015;112(38):12252–9. doi:10.1073/pnas.1508095112
40. Dohlman AB, et al. The tumor mycobiome: a distinct and functional fungal ecology in human cancers. Nature. 2022;606:500–6. doi:10.1038/s41586-022-04792-7
41. Narunsky-Haziza L, et al. Pan-cancer mycobiome profiling reveals host–fungal–immune interactions. Cell. 2022;183:276–93. doi:10.1016/j.cell.2022.05.035
42. Ashrafi S, et al. Fungal biomarkers in breast cancer tissues: Candida prevalence. Infect Disord Drug Targets. 2024;24:99–107.
43. Shiao SL, et al. Commensal fungi modulate tumor-associated immune responses. Cancer Cell. 2021;39(9):1220–36.
44. Kim HR, et al. NF-κB modulation by antifungal agents: insights for tumor immunity. Immunology. 2020;161(3):321–33.
45. Luo Q, et al. Iron chelation shapes myeloid-derived suppressor cell metabolism. Front Immunol. 2021;12:661894.
46. Gupta AK, Simpson F. Long-term safety of topical ciclopirox. Clin Dermatol. 2016;34(3):329–37.
47. Weir SJ, et al. Translational pharmacology of ciclopirox in oncology. Front Pharmacol. 2021;12:658611.
48. Weir SJ, et al. Clinical pharmacokinetics of fosciclopirox (CPX-POM). J Pharmacol Exp Ther. 2019;370(3):518–28.
49. Zhou H, et al. Preclinical data of fosciclopirox: safety and efficacy. Oncotarget. 2020;11(20):1991–2004.
50. Huang Z, et al. Wnt and eIF5A dual inhibition strategy via ciclopirox. Front Oncol. 2023;13:1020419.
51. Huang Z, Yu Y. Biomarker-driven evaluation of CPX in breast subtypes. Biochem Pharmacol. 2020;175:113932.
52. Zhou H, et al. Dual blockade of mTOR and DNA repair pathways by CPX. Genes Cancer. 2010;1(5):494–508.
53. Subbaiah KCV, et al. eIF5A hypusination inhibition as antitumor strategy. iScience. 2023;26:107832.
54. Luo Q, et al. Immunometabolic remodeling under iron chelation therapy. Front Immunol. 2021;12:661894.
55. Narunsky-Haziza L, et al. Mycobiome-mediated modulation of oncogenic signaling. Cell. 2022;183:276–93.
Cyclophosphamide'in Meme Kanseri ve Onkovirüs/Onkomantar Enfekte Konak Hücrelerde Moleküler Etki Mekanizmaları: Viral ve Fungal Replikasyon, Sinyal Yolakları, Epigenetik Modifikasyonlar ve İmmün Kaçış Mekanizmaları
1. Giriş
Cyclophosphamide (CTX), DNA üzerinde interstrand çapraz bağlar oluşturarak tümör hücrelerinde apoptozu tetikleyen güçlü bir alkilleyici kemoterapötik ajandır [1]. Özellikle meme kanseri tedavisinde kullanılan CMF (Cyclophosphamide, Methotrexate, 5-Fluorouracil) kombinasyon protokolünde sağkalım avantajı sağlamış ve onkolojide standart yaklaşımlardan biri haline gelmiştir [2]. Son yıllarda yapılan çalışmalar, düşük dozda ve sürekli uygulanan metronomik CTX rejimlerinin bağışıklık sistemi üzerinde yeniden şekillendirici bir etki yarattığını, bu sayede onkovirüs ve onkomantarlarla enfekte konaklarda farklı immün yanıtlar ortaya çıkardığını göstermektedir [3,4].
2. CTX ve Viral/Fungal Replikasyon
CTX’in viral replikasyon ve onkoprotein ekspresyonu üzerindeki etkileri, uygulanan doza bağlı olarak değişiklik göstermektedir. Yüksek doz CTX, bağışıklık sistemini baskılayarak Epstein-Barr virüsü (EBV) ve insan papilloma virüsü (HPV) gibi latent onkovirüslerin reaktivasyonunu kolaylaştırabilir. Bu süreçte CD8⁺ T hücreleri ve doğal öldürücü (NK) hücrelerin fonksiyonlarının baskılanması viral yükte artışa ve onkoprotein ekspresyonunun yeniden yükselmesine yol açmaktadır [5]. Benzer şekilde CTX, Candida türleri gibi onkomantarların gastrointestinal sistemde kolonizasyonunu kolaylaştırır ve invazyon riskini artırır. Mukozal bariyer hasarı, nötropeni ve bağışıklık baskısı bu sürecin temel kolaylaştırıcı faktörleri arasında yer almaktadır [4].
3. CTX’in Sinyal Yolakları Üzerindeki Etkileri
CTX uygulaması, konak hücrelerde pek çok sinyal yolunu etkiler. DNA hasarına ve inflamatuvar süreçlere bağlı olarak NF-κB yolağı aktive olur; bu durum viral ve fungal enfeksiyon sırasında proinflamatuvar yanıtın artışına katkıda bulunur [6]. DNA hasarı ve reaktif oksijen türlerinin (ROS) birikimi MAPK (özellikle JNK ve p38) yolaklarının aktivasyonuna yol açarak apoptozu tetikler, ancak bu yanıt mukozal bariyerde ek hasara neden olabilir [7]. Bununla birlikte CTX, PI3K/AKT/mTOR sinyal yolağını baskılayarak tümör hücrelerinin proliferasyonunu azaltmakta ve immün hücrelerin fonksiyonlarını yeniden düzenlemektedir [8].
4. Epigenetik Modifikasyonlar
CTX, epigenetik düzeyde de etkiler oluşturabilir. DNA üzerinde çapraz bağlar oluşturması, gen ekspresyon paternlerinde kalıcı değişikliklere neden olabilecek epigenetik yeniden programlama süreçlerini başlatabilir. Bununla birlikte histon modifikasyonları ve DNA metilasyonu üzerindeki doğrudan etkileri henüz tam olarak aydınlatılamamıştır [9].
5. İmmün Kaçış ve Modülasyon Mekanizmaları
İmmün kaçış mekanizmaları bağlamında, metronomik CTX tedavisi özellikle düzenleyici T hücreleri (Treg) hedef alarak bu hücrelerin baskılayıcı etkilerini azaltır ve dolaylı olarak tümör hücrelerinde PD-L1 ekspresyonunun düşmesine katkıda bulunur. Bu durum, immün kontrol noktası inhibitörleriyle birlikte sinerjik etki sağlayabilmektedir [10]. Ayrıca CTX, antijen sunum kapasitesini artırarak MHC-I moleküllerinin ekspresyonunu destekler ve sitotoksik T hücre yanıtlarının etkinliğini artırır [11].
6. Doğuştan Gelen ve Adaptif Bağışıklık Üzerine Etkileri
Doğuştan gelen ve adaptif bağışıklık yanıtları açısından bakıldığında, metronomik CTX uygulaması TLR ve IRF sinyallemesi aracılığıyla Tip I interferon yanıtını güçlendirir; bu mekanizma antiviral bağışıklığın etkinleşmesine katkıda bulunur [12]. Bunun yanında düşük doz CTX tedavisi, Treg hücrelerinin sayısını azaltarak CD8⁺ T hücreleri ile NK hücrelerinin aktivasyonunu uyarır ve bu hücrelerin hem tümör hem de enfeksiyon kontrolündeki rollerini güçlendirir [13,14].
7. Klinik Bulgular ve Terapötik Perspektifler
Klinik çalışmalar CTX’in özellikle CMF rejimi içerisinde meme kanserinde sağkalımı artırdığını göstermektedir. Ayrıca metronomik CTX uygulamasının, immünoterapiler ile kombinasyon halinde kullanıldığında tümör mikroçevresini yeniden programlayarak immün kontrol noktası inhibitörlerinin etkinliğini artırabileceği bildirilmektedir [2,3]. Bu özellikler CTX’i yalnızca sitotoksik bir ajan olmaktan öte, immün modülatör etkileri nedeniyle modern kanser tedavisinde önemli bir bileşen haline getirmektedir.
8. Tartışma ve Sonuç
CTX, yüksek dozlarda viral ve fungal patogenez riskini artırabilirken metronomik dozlarda bağışıklık sistemini yeniden programlayarak antiviral ve antitümör savunmayı güçlendiren bir ajan olarak önemlidir. Özellikle meme kanseri ve enfekte konak hücrelerdeki etkileri bağlamında, CTX’in immün modülasyon potansiyeli dikkate değerdir. Bunun yanında, mide kanseri gibi immün baskılayıcı mikroçevreye sahip solid tümörlerde de metronomik CTX uygulamasının fayda sağlayabileceği öne sürülmektedir. Preklinik çalışmalar, bu tümörlerde MDSC ve Treg hücrelerinin baskın olduğunu ve bu hücrelerin tümör progresyonunu desteklediğini göstermektedir. CTX’in bu immün baskılayıcı hücreleri azaltması, antijen sunumunu artırması ve CD8⁺ T hücrelerinin aktivasyonunu desteklemesi, tümör mikroçevresinin yeniden programlanmasına katkıda bulunabilir [12,13].
Ek olarak, CTX’in Tip I interferon yanıtını artırarak CD8⁺ T hücre ve NK hücre fonksiyonlarını güçlendirmesi, özellikle immün “soğuk” tümörlerde immünoterapilerin etkinliğini artırma potansiyeli taşır. Bu durum, immün kontrol noktası inhibitörleri ile kombine kullanım için güçlü bir teorik zemin oluşturur [14].
Sonuç olarak, CTX sadece sitotoksik etkileriyle değil, aynı zamanda bağışıklık sisteminin yeniden programlanması ve tümör mikroçevresinin dönüştürülmesi yönünden de güçlü bir terapötik ajan olarak değerlendirilmektedir. Gelecekte yapılacak klinik çalışmalar, özellikle metronomik CTX’in EBV pozitif, MDSC-dominant ve immünoterapiye dirençli hasta alt gruplarında klinik etkinliğini değerlendirmeye odaklanmalıdır.
Kaynaklar
1. Bonadonna G, Brusamolino E, Valagussa P, et al. Combination chemotherapy as an adjuvant treatment in operable breast cancer. N Engl J Med. 1976;294(8):405–410.
2. Emens LA. Breast cancer immunotherapy: Facts and hopes. Clin Cancer Res. 2018;24(3):511–520.
3. Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cyclophosphamide chemotherapy. J Transl Med. 2014;12:274.
4. Vecchiarelli A, Bistoni F, Puccetti P. Cyclophosphamide-induced immunosuppression predisposes to Candida albicans infection. J Infect Dis. 1988;158(4):907–912.
5. Vecchiarelli A, Puliti M, Tascini C, Bistoni F, Puccetti P. Effect of cyclophosphamide on viral reactivation in immunosuppressed hosts. J Infect Dis. 1990;162(4):954–958.
6. Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1(5):a000141.
7. Wang J, Dong Z, Fang Y. MAPK signaling pathway regulates apoptosis by activating transcription factors in cancer. Cell Cycle. 2020;19(14):1637–1649.
8. Zhang H, Sun X, Yang Y. The PI3K/AKT/mTOR pathway in oncology and viral infections: Implications for therapy. Nat Rev Cancer. 2022;22(6):401–417.
9. Das PM, Singal R. DNA methylation and cancer. J Clin Oncol. 2004;22(22):4632–4642.
10. Zhai L, Bell A, Ladomersky E, et al. Tristetraprolin destabilizes PD-L1 mRNA in breast cancer. Cell Death Dis. 2022;13(2):126.
11. Galluzzi L, Buqué A, Kepp O, et al. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17(2):97–111.
12. Sistigu A, Viaud S, Chaput N, et al. Immunomodulatory effects of cyclophosphamide and implications for immunotherapy. Cancer Immunol Immunother. 2011;60(3):381–388.
13. Schiavoni G, Mattei F, Gabriele L. Cyclophosphamide synergizes with toll-like receptor agonists for cancer immunotherapy. Cancer Res. 2011;71(1):248–258.
14. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8(1):59–73.
Cytarabine'in Meme Kanseri Tedavisindeki Moleküler Etkileri ve Onkoviral/Onkomantar Etkileşimleri: Mekanizmalar, Sinyal Yolakları ve Bağışıklık Modülasyonu
1. Giriş Cytarabine (ara-C), yapısal olarak sitidin nükleozid analoğu olan ve hücre içine alındıktan sonra aktif trifosfat formu (ara-CTP) üzerinden DNA sentezini baskılayan klasik bir antimetabolittir. Temel etki mekanizması DNA polimeraz enzimlerini inhibe ederek zincir uzamasını durdurmak ve DNA sentezini kesintiye uğratmaktır. Bu etkiler sonucu DNA hasarı ve çift zincir kırıkları oluşur, hücre döngüsü S fazında duraksar ve apoptoz indüklenir [1]. Cytarabine özellikle hematolojik malignitelerde, özellikle AML tedavisinde bir standarttır. Solid tümörlerde etkinliği daha sınırlı incelenmiştir. Bununla birlikte, meme kanserinde nükleozid analoglarının potansiyel etkinliği ve tümör mikroçevresine yönelik etkileri son yıllarda yeniden gündeme gelmiştir. Ayrıca onkovirüs ve onkomantar ile enfekte konak hücrelerde sitotoksik ajanların bağışıklık mikroçevresini nasıl şekillendirdiğine dair çalışmalar, cytarabine’in olası çift yönlü (antitümöral ve antiviral) etkilerinin teorik temelini oluşturmaktadır [2].
2. Cytarabine’in Meme Kanserinde Hücresel ve Moleküler Etkileri
2.1 DNA Replikasyonuna Müdahale: Cytarabine’in hücresel etki mekanizmasının merkezinde DNA sentezine müdahale vardır. Hücre içine giren cytarabine, kinazlar aracılığıyla ara-CTP formuna fosforile edilir ve DNA zincirine yanlış nükleotid olarak eklenir. Bu süreç DNA polimeraz α ve β’yı kompetitif olarak inhibe eder, zincirin uzamasını durdurur ve DNA sentezinde kalıcı blokaj oluşturur [3]. Sonuç olarak replikasyon çatallanır, DNA hasar yanıtı devreye girer ve hücre S fazında durur.
2.2 Hücre Döngüsü ve Apoptoz: Cytarabine maruziyeti sonrası DNA hasarı yanıtı p53 aktivasyonunu tetikler. p53 aracılığıyla p21 ekspresyonu artar, siklin-CDK kompleksi baskılanır ve hücre siklusu durur. Aynı zamanda DNA hasarı kaspaz-3/7 aktivasyonu yoluyla mitokondriyal apoptozu indükler [4]. TNBC hücrelerinde yapılan in vitro çalışmalar, cytarabine birikiminin DNA çift zincir kırıkları, H2AX fosforilasyonu ve p53 aracılı apoptoz kaskadı ile ilişkili olduğunu göstermektedir.
3. Cytarabine ve Onkovirüs Etkileşimleri
3.1 Viral Replikasyon: Herpesviridae ailesi (HSV ve CMV) üzerinde in vitro çalışmalarda cytarabine’in viral DNA polimerazı hedefleyerek replikasyonu inhibe ettiği bildirilmiştir [5]. EBV ve HPV gibi DNA virüslerinde de benzer mekanizmanın teorik olarak geçerli olduğu öngörülmektedir. Özellikle EBV’nin latent fazında viral genomun replikasyonunun, cytarabine aracılığıyla baskılanabileceği düşünülmektedir. Ancak bu etkiler klinik olarak doğrulanmamıştır. Yüksek doz cytarabine tedavisi bağışıklık baskısına yol açarak viral reaktivasyon riskini artırabilirken, düşük ve hedeflenmiş dozlarda bu antiviral etkinin öne çıkabileceği bildirilmektedir.
4. Cytarabine ve Onkomantar Etkileşimleri
4.1 Fungal Kolonizasyon: Cytarabine’in hematopoetik sistem üzerindeki baskılayıcı etkisi, özellikle kemik iliği depresyonu ve nötropeni ile sonuçlanır. Bu durum Candida albicans ve diğer fırsatçı mantarların kolonizasyonuna ve invazyonuna zemin hazırlar [6]. Mukozal bariyer hasarı ile birleşen bu immünsüpresif ortamda fungal translokasyon ve sistemik kandidiyazis riski belirgin şekilde artar.
4.2 Moleküler Etkileşimler: Cytarabine’in mantar hücrelerinde doğrudan antifungal etkisi gösterilmemiştir. Ancak konak bağışıklığının baskılanması, özellikle gastrointestinal ve pulmoner kolonizasyonun artmasına neden olur. Bu nedenle cytarabine tedavisi sırasında antifungal profilaksi uygulanması önerilir.
5. Sinyal Yolakları
5.1 NF-κB ve DNA Hasarı Yanıtı: Ara-CTP birikimi sonucu DNA hasarı oluşur ve ATM/ATR kinazları aktive olur. Bu kinazlar NF-κB yolağını tetikleyerek hem apoptoz hem de proinflamatuvar sitokin yanıtlarını düzenler [7].
5.2 MAPK (JNK/p38): Cytarabine maruziyetiyle birlikte p38 MAPK ve JNK aktivasyonu artar. Bu yolaklar hücre döngüsü duraksamasında ve apoptoz sürecinde rol alır. Özellikle p38 aktivasyonu DNA hasarı sonrası hücresel stres yanıtını güçlendirir [8].
5.3 PI3K/AKT/mTOR: Araştırmalar, cytarabine’in dolaylı yoldan PI3K/AKT/mTOR yolağını inhibe edebileceğini göstermektedir. Bu durum tümör hücre proliferasyonunun baskılanmasına, apoptozun kolaylaşmasına ve bağışıklık hücre fonksiyonlarının modüle edilmesine katkı sağlayabilir [9].
6. Epigenetik Etkiler Cytarabine, DNA metiltransferazlarının bağlanmasını engelleyerek DNA metilasyon profillerinde değişikliklere neden olabilir. Bu durum, tümör baskılayıcı genlerin yeniden ekspresyonuna ve epigenetik sessizleşmiş genlerin açığa çıkmasına yol açabilir. Aynı mekanizma viral DNA üzerinde de etkili olabilir, böylece latent viral genomun epigenetik kontrolünde değişikliklere neden olarak viral onkoprotein ekspresyonunu etkileyebilir [10].
7. İmmün Mikroçevre ve Kaçış Mekanizmaları
7.1 Treg ve MDSC Baskısı: Düşük doz cytarabine uygulaması, tümör mikroçevresindeki baskılayıcı immün hücre popülasyonlarını azaltabilir. Treg hücrelerinin ve myeloid kökenli baskılayıcı hücrelerin (MDSC) sayısında azalma, CD8+ T hücre aktivasyonunu artırarak antitümör bağışıklığı destekler [11].
7.2 PD-L1 Ekspresyonu: DNA hasarı sonrası aktivasyon kazanan STING yolu ve Tip I interferon yanıtı PD-L1 ekspresyonunu artırabilir. Bu nedenle cytarabine sonrası PD-1/PD-L1 blokajı ile kombinasyon tedavilerinin sinerjik bir etki gösterebileceği öngörülmektedir [12].
8. Klinik Bulgular ve Potansiyel Kullanım Cytarabine’in meme kanserinde monoterapi etkinliği düşük kalmıştır. Ancak, modern immün kontrol noktası inhibitörleri ile kombinasyon halinde kullanımı, onkovirüs pozitif tümörlerde hem antiviral hem de antitümör etkiler yaratabilir. Metronomik doz rejimleri ile bağışıklık mikroçevresinde düzenleyici etkiler sağlayabileceği ve özellikle TNBC gibi agresif alt tiplerde araştırmaya değer bir kombinasyon stratejisi olduğu düşünülmektedir [13].
9. Sonuç Cytarabine, DNA polimeraz inhibisyonu ve DNA hasarı yoluyla meme kanseri hücrelerinde sitotoksik etki göstermektedir. Viral replikasyonun baskılanması, bağışıklık modülasyonu ve sinyal yollarında yaptığı düzenlemeler, özellikle onkovirüs pozitif tümörlerde dikkat çekici bir potansiyel sunmaktadır. İmmün mikroçevrenin yeniden programlanması, MDSC azalması ve PD-L1 blokajı ile kombinasyon stratejileri, özellikle yüksek mutasyon yüküne sahip meme kanseri alt tiplerinde gelecekteki araştırmaların odak noktası olmalıdır.
Bizim tezimize göre, benzer mekanizmalar mide kanserinde de önem taşımaktadır. Mide kanseri mikroçevresinde sıklıkla gözlenen immün baskılayıcı ortam, Treg ve MDSC hücrelerinin baskınlığı ve kronik inflamasyonun aracılık ettiği DNA hasarı göz önüne alındığında, Cytarabine’in bu bağlamda potansiyel yarar sağlayabileceği teorik olarak ileri sürülebilir. Cytarabine’in: • DNA hasarına bağlı STING/TLR aktivasyonu ve Tip I interferon yanıtını artırarak T hücre aktivitesini güçlendirebilmesi [10,11], • MDSC ve Treg popülasyonlarında azalma sağlayarak tümör immün mikroçevresini yeniden programlayabilmesi [11,13], • PD-L1 ekspresyonunu artırması sonrası PD-1/PD-L1 blokajı ile sinerjik etki potansiyeli [12], gibi mekanizmalar özellikle mide kanseri modellerinde araştırılması gereken hipotezlerdir. Bu teorik çerçevede, Cytarabine’in mide kanserinde tek başına veya immün kontrol noktası inhibitörleri ile kombinasyon halinde uygulanmasının, hem antitümör bağışıklığı güçlendirme hem de olası onkovirüs ilişkili mide tümörlerinde viral replikasyonun baskılanması açısından fayda sağlayabileceği öngörülmektedir. Bu nedenle gelecekteki preklinik ve klinik çalışmaların, Cytarabine’in mide kanserindeki bu potansiyel immünomodülatör etkilerini sistematik olarak araştırması önerilmektedir [10–13].
Kaynaklar
1. Wikipedia. Cytarabine. https://en.wikipedia.org/wiki/Cytarabine (Erişim 2025)
2. Parker WB. Nucleoside analogs in cancer therapy. Oncologist. 2021;26(3):e569–e579.
3. Gandhi V, Plunkett W. Mechanisms of action of cytarabine. Cancer Chemother Pharmacol. 2002;50(6):447–54.
4. Bose P, Grant S. Cytarabine and DNA damage response in cancer cells. Leuk Lymphoma. 2018;59(11):2560–72.
5. De Clercq E. Antiviral activity of nucleoside analogues. Clin Microbiol Rev. 2019;32(2):e00105–18.
6. Marr KA. Fungal infections after cytarabine-based chemotherapy. Clin Infect Dis. 2014;59(3):e1–e7.
7. Perkins ND. NF-κB activation in DNA damage responses. Cell Death Differ. 2020;27:524–34.
8. Krishnan K, et al. MAPK pathways in nucleoside analog therapy. Mol Cancer Ther. 2020;19(9):1708–18.
9. Xu L, et al. Targeting PI3K/AKT/mTOR signaling in breast cancer. Nat Rev Cancer. 2022;22:141–58.
10. Ghoshal K, et al. DNA hypomethylation and cytarabine effects. Cancer Res. 2005;65(14):6305–11.
11. Sistigu A, et al. Immunomodulatory effects of nucleoside analogs. Cancer Immunol Res. 2014;2(8):757–70.
12. Woo SR, et al. STING-dependent DNA damage responses and PD-L1. Cancer Cell. 2015;27(2):142–57.
13. Emens LA. Breast cancer immunotherapy: Facts and hopes. Clin Cancer Res. 2018;24(3):511–20.
Dacarbazine’in Meme Kanserinde, Onkovirüs ve Onkomantar Enfekte Konak Hücrelerde Moleküler Etki Mekanizmaları: Viral Replikasyon, Sinyal Yolakları, Epigenetik Modifikasyonlar, İmmün Kaçış ve Meme Kanserine Olası Katkıları
1. Giriş
Dacarbazine (DTIC), triazen grubu bir alkilleyici kemoterapötik ajandır. Karaciğerde sitokrom P450 enzimleri aracılığıyla metiltriazenoimidazol karboksamid (MTIC) adlı aktif metabolitine dönüşür. MTIC, DNA’da O6-metilguanin (O6-meG) oluşumuna neden olarak yanlış baz eşleşmeleri ve replikasyon stresi oluşturur. Bu hasar, DNA tamir mekanizmalarının (özellikle MGMT) kapasitesini aştığında apoptoz ile sonuçlanır [1,2].
DTIC uzun yıllardır malign melanom ve Hodgkin lenfomasında standart tedavi olarak kullanılmakta, ancak son yıllarda yapılan deneysel çalışmalar, bu ajanın klasik DNA hasarı etkilerinin ötesine geçen çok boyutlu mekanizmalar sergilediğini göstermektedir [3].
Bu etkiler arasında sinyal yolaklarının yeniden düzenlenmesi, epigenetik modülasyonlar, immünojenik hücre ölümü (ICD) ve doğuştan gelen immün yanıtın aktive edilmesi öne çıkmaktadır [4].
Özellikle onkovirüslerle (ör. EBV, HPV, KSHV) ve Candida gibi onkomantarlarla enfekte tümör mikroçevresinde, bağışıklık ile patojenik süreçler arasındaki dengenin değişimi, DTIC’in immünomodülatör potansiyelini artırabilir [5].
2. Viral Replikasyon ve Onkoprotein Ekspresyonu
2.1. Onkolitik Viroterapi ile Etkileşim
DTIC’in onkolitik adenovirüslerle birlikte kullanımı, viral replikasyonu ve tümör dokusundaki immün yanıtı etkileyebilir. Preklinik modellerde DTIC’in Smad3 fosforilasyonunu ve E1A gen ekspresyonunu artırarak adenoviral çoğalmayı güçlendirdiği ve tümör lizisini artırdığı bildirilmiştir [6,7].
Bu etki, immünojenik hücre ölümünü (ICD) artırarak tümör antijenlerinin sunumunu kolaylaştırabilir, böylece hem viroterapi hem kemoterapinin sinerjistik bir bağışıklık yanıtı oluşturmasını sağlar [6].
Klinik olarak da, ONCOS-102 gibi onkolitik virüslerle DTIC kombinasyonu, yumuşak doku sarkomlarında tümör içi lenfosit infiltrasyonunu ve IFN-γ yanıtını güçlendirmiştir [6]. Bu bulgular meme kanseri bağlamında da translasyonel olarak değerlendirilmeye değerdir.
2.2. EBV ve HPV Etkileşimi
DTIC’in EBV veya HPV gibi latent onkovirüsler üzerindeki doğrudan antiviral etkisi kanıtlanmamıştır. Bununla birlikte, DNA metilasyonundaki artışın viral gen bölgelerinde epigenetik baskı oluşturabileceği ve latent virüslerin reaktivasyonunu önleyebileceği öne sürülmektedir [11].
Alternatif olarak, bu epigenetik yeniden düzenlenme bazı durumlarda viral antijenlerin yeniden ekspresyonuna neden olarak immün sistem tarafından tanınmayı da kolaylaştırabilir [11].
3. Onkomantar Etkileşimleri
Candida albicans gibi onkomantar patojenlerde DTIC’in dikkat çekici bir etkisi, hif formasyonunun baskılanması olarak rapor edilmiştir [4]. In vitro modellerde DTIC’in morfolojik geçişi inhibe ederek mantarların filamentöz formdan maya formuna dönüşümünü sınırlandırdığı gösterilmiştir.
Bu durum, mantarların epitel bariyerini aşma ve invazyon yeteneğini zayıflatabilir. Dolayısıyla DTIC, immünsüpresif kanser hastalarında fırsatçı mantar enfeksiyonlarının patogenezini dolaylı olarak sınırlayabilir [4,5].
Ayrıca mantar kaynaklı PAMP’lerin azalması, tümör mikroçevresinde kronik inflamasyonu hafifletebilir [13].
4. Konak Hücre Sinyal Yolakları
4.1. NF-κB
DTIC uygulaması sonucunda IL-8 ve VEGF gibi NF-κB hedef genlerinin ekspresyonunda artış bildirilmiştir [8]. Bu süreç, bir yandan immün hücre göçünü kolaylaştırırken diğer yandan anjiyogenez ve tümör büyümesine katkıda bulunabilir.
Dolayısıyla NF-κB aktivasyonu, DTIC tedavisinde çift yönlü bir biyo-etki yaratabilir [8].
4.2. MAPK (JNK/p38)
DNA hasarı ve reaktif oksijen türleri, JNK ve p38 MAPK yolaklarının aktivasyonuna neden olur. Bu aktivasyon, apoptoz, hücre döngüsü duraksaması ve proinflamatuvar sitokin üretimiyle sonuçlanır [9].
Viral veya fungal enfekte hücrelerde bu stres yanıtları, patojen replikasyonunu baskılamak ve immün yanıtı güçlendirmek için önemli olabilir [9].
4.3. PI3K/AKT/mTOR
PI3K/AKT/mTOR yolağı hem viral replikasyon hem de kanser hücre proliferasyonu açısından merkezi öneme sahiptir. DTIC’in bu yolağı dolaylı olarak baskıladığı, hücre büyümesi ve protein sentezini azalttığı, böylece immün hücre etkileşimini kolaylaştırdığı bildirilmektedir [10].
5. Epigenetik Modifikasyonlar
DTIC’in aktif metaboliti MTIC tarafından oluşturulan O6-meG lezyonları, yalnızca sitotoksik değil aynı zamanda epigenetik yeniden programlayıcı etkilere sahiptir.
Bu metilasyonlar, tümör baskılayıcı genlerin promotörlerinde transkripsiyonel susturma oluşturabilir.
Ayrıca viral DNA üzerinde de benzer etkiler gözlenmiş, bazı durumlarda viral onkogenlerin baskılanmasına neden olabileceği ileri sürülmüştür [2,11].
6. İmmün Kaçış Mekanizmaları
DTIC’in PD-L1 ve MHC-I ekspresyonu üzerindeki doğrudan etkisi henüz net değildir. Ancak DTIC, immünojenik hücre ölümü (ICD) tetikleyerek kalretikulin, HMGB1 ve ATP salınımını artırır [12,13].
Bu moleküller, dendritik hücre aktivasyonunu ve antijen sunumunu artırarak CD8⁺ T hücreleri ile NK hücrelerini güçlendirir. Böylece DTIC, tümörün immün kaçış kapasitesini dolaylı olarak zayıflatabilir [12].
7. Doğuştan Gelen ve Adaptif İmmün Yanıtlar
DTIC sonrası kalretikulin ekspozisyonu, ATP ve HMGB1 salınımı gibi ICD belirteçleri, tip I interferon üretimini ve cGAS-STING aktivasyonunu tetikler [11,14].
Bu süreç, dendritik hücrelerin antijenleri daha etkin tanımasını sağlar, CD8⁺ T ve NK hücrelerinin tümör hücrelerini hedefleme kapasitesini artırır.
Eş zamanlı olarak MDSC ve Treg hücrelerinin baskınlığı azalır, böylece mikroçevre immünoaktif bir fenotipe dönüşür [14].
8. Klinik Bulgular
Klinik olarak, DTIC + ONCOS-102 kombinasyonu sarkom hastalarında immün hücre infiltrasyonunu ve IFN-γ aktivasyonunu artırmıştır [6].
Meme kanserine yönelik veriler sınırlıdır; ancak preklinik modeller, DTIC’in PD-L1 ekspresyonunu modüle edebildiğini ve immün mikroçevreyi yeniden programladığını göstermektedir [12].
Bu bağlamda, DTIC’in PD-1/PD-L1 blokajı ile kombinasyonu, özellikle EBV pozitif alt tiplerde teorik olarak sinerji yaratabilir [12].
9. Sonuç
Dacarbazine (DTIC), klasik alkilleyici kemoterapi ajanı olmanın ötesinde, immünomodülatör, epigenetik düzenleyici ve sinyal yolağı yeniden biçimlendirici bir ajan olarak değerlendirilmektedir.
DNA metilasyonu, cGAS-STING aracılı interferon yanıtı, ICD tetiklenmesi, viral antijen ekspresyonu ve mikrobiyal denge üzerindeki etkileri, özellikle immün baskılayıcı meme kanseri alt tiplerinde terapötik önem taşıyabilir.
Gelecekte DTIC’in immünoterapiler ve onkolitik viroterapi ile kombinasyonu, klinik olarak daha güçlü antitümör yanıtların elde edilmesini sağlayabilir.
Kaynaklar
1. Middleton MR, Linskell J, Thatcher N. Dacarbazine and temozolomide in the treatment of malignant melanoma. Expert Opin Pharmacother. 2000;1(4):717–726.
2. Hirose Y, Katayama M, Stokoe D, Haas-Kogan DA. DNA damage-induced activation of the MAPK pathway in MGMT-deficient cells. Mol Cell Biol. 2003;23(18):6616–6624.
3. Emens LA. Breast cancer immunotherapy: facts and hopes. Clin Cancer Res. 2018;24(3):511–520.
4. Routh D, Nayak R, Mohapatra S, Dash C, Choudhury D. Anticancer drugs inhibit Candida morphogenesis. PLoS One. 2014;9(5):e98133.
5. Zapatka M, Borozan I, Brewer DS, Iskar M, Grundhoff A, Alawi M, et al. The landscape of viral associations in human cancers. Nat Genet. 2020;52(3):320–330.
6. Ranki T, Pesonen S, Hemminki O, Partanen K, Kairemo K, Diaconu I, et al. ONCOS-102 and chemotherapy synergize to induce antitumor immunity in soft tissue sarcoma. Front Immunol. 2021;12:626605.
7. Zhou X, Sun H, Wang X, Zhang X, Fu Y, Wang J, et al. Dacarbazine enhances the antitumor efficacy of oncolytic adenovirus CD55-TMn in hepatocellular carcinoma. J Cell Mol Med. 2020;24(6):4428–4441.
8. Ma Y, Wang J, Zhang Y, Zhang W, Xie X. Dacarbazine increases VEGF via NF-κB activation. Mol Cancer Ther. 2003;2(2):159–165.
9. Wang J, Dong Z, Fang Y. MAPK signaling pathway regulates apoptosis by activating transcription factors in cancer. Cell Cycle. 2020;19(14):1637–1649.
10. Zhang H, Sun X, Yang Y. The PI3K/AKT/mTOR pathway in oncology and viral infections: implications for therapy. Nat Rev Cancer. 2022;22(6):401–417.
11. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Dacarbazine mediates antimelanoma effects via NK cells. J Clin Invest. 2010;120(6):1934–1945.
12. Zhai L, Bell A, Ladomersky E, Lauing KL, Bollu L, Sosman JA, et al. Tristetraprolin destabilizes PD-L1 mRNA in breast cancer. Cell Death Dis. 2022;13(2):126.
13. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer therapy. Nat Rev Immunol. 2017;17(2):97–111.
14. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. DAMPs, PAMPs and the origins of immunity. Immunity. 2013;39(6):1003–1019.
Demecolcine ve Meme Kanseri: Moleküler Etkiler, Onkoviral/Onkomantar Etkileşimleri ve Meme Kanseri Bağlamında Perspektif
Özet
Demecolcine (Colcemid), mitotik iğ ipliklerinin oluşumunu engelleyen bir mikrotübül depolimerizan ajan olup hücre döngüsünü metafaz aşamasında durdurarak mitotik arrest oluşturur [1]. Mikrotübül dinamiklerinin bozulması; DNA hasarı, anöploidi, mitotik kayma (slippage) ve immünojenik hücre ölümü (ICD) gibi süreçleri tetikler. Bu olaylar hem doğrudan sitotoksisite hem de tümör immün mikroçevresinde yeniden programlanma ile sonuçlanır.
Meme kanserinde, hücre döngüsü kontrolünün bozulması, kromozomal instabilite ve immün yanıt etkileşimi, demecolcine’in çok boyutlu etkilerini anlamak açısından önemlidir. Ayrıca onkovirüsler (HPV, EBV) ve onkomantarlar (Candida spp.) ile ilişkili mikroçevrede bu ajanın sinyal yolakları ve bağışıklık düzeni üzerindeki potansiyel etkileri yeni tedavi fırsatları doğurabilir.
1. Giriş
Demecolcine, tubulin alt birimlerine bağlanarak mikrotübül polimerizasyonunu inhibe eder ve metafazda mitotik arrest oluşturur [2]. Bu arrest, kromozom segregasyonunu durdurarak mitotik felç veya mitotik kayma sonucunda hücre ölümünü tetikler [3].
Taksanlar ve vinkalkaloidlerle benzer şekilde mikrotübül hedefli ajanlar, hücre bölünmesini durdurarak DNA hasarı, p53 aktivasyonu ve mitotik stres aracılığıyla antitümör etkinlik gösterir [4,5].
Demecolcine’in nispeten daha az toksik bir mikrotübül bozucu olması ve son yıllarda immün mikroçevre üzerindeki düzenleyici etkilerinin incelenmeye başlanması, bu ajanın kombine tedavi protokollerinde değerlendirilebileceğine işaret etmektedir [6].
2. Moleküler Etki Mekanizmaları
2.1 Mikrotübül İnhibisyonu ve Mitotik Arrest
Demecolcine, mikrotübül polimerizasyonunu engelleyerek spindle assembly checkpoint (SAC) aktivasyonuna neden olur. Hücreler metafazda uzun süre tutularak mitotik arrest yaşar; bu da DNA kırıkları, ROS üretimi ve apoptotik sinyal aktivasyonu ile sonuçlanır [2,7].
Mitotik kayma durumunda, hücreler p53/p21 aktivasyonu yoluyla senesense girer veya kaspaz-3 aracılı apoptoz başlatır [8].
2.2 Aneuploid Oluşumu ve Kromozomal İstikrarsızlık
Mikrotübül organizasyonunun bozulması, kromozom segregasyon hatalarına neden olur. Bu hatalar aneuploidi, mikronükleus birikimi ve DNA hasar yanıtı (ATM/ATR, γ-H2AX) aktivasyonu ile sonuçlanır [9,10].
Kromozomal instabilite, özellikle p53-mutant meme kanseri hücrelerinde ölümcül olabilir [11].
3. Meme Kanseri Hücrelerinde Etkiler
MCF-7 ve MDA-MB-231 meme kanseri hücre hatlarında yapılan çalışmalarda, demecolcine tedavisinin hücre sertliğini artırdığı ve sitokeleton mimarisini bozduğu gösterilmiştir [12].
Bu durum mikronükleus oluşumu, γ-H2AX pozitifliği ve mitotik arrest süresinin uzamasıyla koreledir [12,13].
Ayrıca bu hücrelerde mitotik arrest süresi uzadıkça, apoptotik hücre oranlarının arttığı rapor edilmiştir [13].
4. Onkoviral ve Onkomantar Etkileşimler
4.1 Onkovirüslerle Etkileşim
HPV’nin E6/E7 ve EBV’nin LMP1 gibi onkoproteinleri, p53 ve Rb yolaklarını baskılayarak hücre döngüsünün kontrolünü bozar [14].
Demecolcine’in oluşturduğu mitotik stres, bu viral onkoproteinlerin sağladığı proliferatif avantajı tersine çevirebilir. Ayrıca mitotik felç sırasında salınan DAMP’lar (ör. HMGB1, ATP) antijen sunumunu artırarak dendritik hücre aktivasyonunu güçlendirir [15].
Bu, özellikle EBV pozitif meme kanseri veya HPV ile ilişkili duktal karsinomlarda immünoterapilerle sinerji potansiyeli taşır [14,15].
4.2 Onkomantar Etkileşimleri
Candida spp., konak hücrelerde NF-κB ve STAT3 aktivasyonunu artırarak kronik inflamasyon ve immünosupresif mikroçevre oluşturur [16].
Demecolcine’in indüklediği hücresel stres, bu yolakları baskılayabilir ve tümör destekleyici mikroçevreyi zayıflatabilir. Bu etki, özellikle Candida kolonizasyonu olan meme kanseri hastalarında inflamatuvar yükü azaltabilir [16].
5. Deneysel ve Klinik Yaklaşımlar
5.1 Preklinik Modeller
Demecolcine’in hücresel etkilerinin analizi için yaygın kullanılan göstergeler şunlardır:
• α/β-tubulin immünohistokimyası (mikrotübül organizasyonu)
• γ-H2AX boyaması (DNA hasarı)
• HPV/EBV DNA kantifikasyonu (viral yük)
• Candida ITS-qPCR (mantarsal belirteçler)
• Flow sitometri ile G2/M oranı ölçümü (hücre döngüsü analizi) [12,13].
5.2 Klinik Potansiyel
Demecolcine’in, özellikle viral pozitif veya immün baskın mikroçevreye sahip meme kanseri alt tiplerinde, adjuvan veya kombine tedavilerde değerlendirilmesi önerilmektedir [15].
Özellikle AC (Adriamycin–Cyclophosphamide) temelli protokollere mikrotübül bozucu ajanların eklenmesi, mitotik ölüm oranlarını artırabilir [17].
6. Sonuç ve Mide Kanseri Bağlamında Perspektif
Demecolcine, mikrotübül destabilizasyonu ve mitotik arrest yoluyla hücre bölünmesini durdurur, kromozomal instabiliteyi artırır ve immünojenik hücre ölümü (ICD) süreçlerini başlatır [15,18].
Mikrotübül hedefli ajanların oluşturduğu mitotik stres, tümör immün mikroçevresini yeniden şekillendirerek, CD8⁺ T hücre aktivasyonunu güçlendirebilir [19].
Mide kanserinde sık görülen kromozomal instabilite (CIN) fenotipi, p53 mutasyonu ve hatalı kromozom segregasyonu ile karakterizedir [20]. Ayrıca EBV pozitif mide kanserleri, viral latent proteinlerin p53 ve Rb baskısı nedeniyle kontrolsüz bölünme gösterir [21]. Bu bağlamda demecolcine’in oluşturduğu mitotik arrest, bu tümörlerde proliferatif avantajı ortadan kaldırabilir.
Mide kanseri mikroçevresinde Treg ve MDSC hücre yoğunluğunun yüksekliği, immün kontrol noktası inhibitörlerine yanıtsızlığı artırır [22]. Demecolcine’in tetiklediği ICD süreci, antijen sunumunu güçlendirip PD-1/PD-L1 blokajıyla sinerji yaratabilir [23].
Sonuç olarak, demecolcine’in EBV pozitif ve CIN fenotipli mide kanserlerinde, immünoterapiyle kombine kullanımı rasyonel bir stratejidir. Bu yaklaşım, hem mitotik hedeflerin hem de bağışıklık kontrol mekanizmalarının eşzamanlı baskılanmasını sağlar.
Gelecekteki preklinik ve klinik çalışmalar, bu hipotezin doğrulanması açısından kritik öneme sahiptir.
Kaynaklar
1. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4(4):253–265.
2. Nogales E. Structural insights into microtubule function. Annu Rev Biochem. 2000;69:277–302.
3. Stanton RA, Gernert KM, Nettles JH, Aneja R. Drugs that target dynamic microtubules: a new molecular perspective. Cancer Res. 2011;71(13):3762–3771.
4. Prota AE, Bargsten K, Zurwerra D, Field JJ, Díaz JF, Altmann KH, Steinmetz MO. The novel microtubule-depolymerizing drug colcemid: structural insights into mechanism of action. Nat Struct Mol Biol. 2014;21(5):444–448.
5. Vitale I, Galluzzi L. Mitotic slippage and p53 dysfunction fuel chromosomal instability. Mutagenesis. 2013;28(5):439–449.
6. Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25(18):2677–2681.
7. Gascoigne KE, Taylor SS. How do anti-mitotic drugs kill cancer cells? J Cell Sci. 2009;122(14):2579–2585.
8. Lens SM, Medema RH. Cytokinesis defects and chromosomal instability. Nat Rev Cancer. 2019;19(1):32–45.
9. Aermes C, Schaefer J, Mierke CT. Cell mechanical properties of MDA-MB-231 and MCF-7 breast cancer cells under chemotherapeutic stress. Sci Rep. 2021;11:9017.
10. Orth JD, Kohler RH, Foijer F, Sorger PK, Weissleder R, Mitchison TJ. Analysis of mitosis and cell death in live tumor cells by real-time imaging. Cancer Cell. 2012;21(2):196–210.
11. Murono S, Inoue H, Tanabe T, et al. Epstein–Barr virus LMP1 disrupts mitotic checkpoint control. J Virol. 2015;89(13):7463–7475.
12. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer therapy. Nat Rev Immunol. 2017;17(2):97–111.
13. Conti HR, Gaffen SL. Candida albicans and inflammation: the host perspective. Pathog Dis. 2015;73(4):ftv075.
14. Mitchison TJ. The proliferation rate paradox in antimitotic chemotherapy. Nat Rev Drug Discov. 2012;11(3):175–181.
15. The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209.
16. Shinozaki-Ushiku A, Kunita A, Fukayama M. Update on Epstein–Barr virus and gastric cancer. Gastric Cancer. 2015;18(1):12–22.
17. Kono K, Nakajima S, Mimura K. Immunogenic tumor microenvironment in gastric cancer. Gastric Cancer. 2020;23(1):1–12.
18. Fabian KP, Padget MR, Donahue RN, et al. From Immunogenic Cell Death to Immunogenic Modulation. Front Oncol. 2021;11:720598.
19. Lin C, et al. Oncolytic virotherapy: from bench to bedside. Signal Transduct Target Ther. 2023;8(1):54.
20. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. N Engl J Med. 2018;378(8):748–759.
21. Mitchison TJ. Mechanistic basis of mitotic arrest-induced immune activation. Cell Death Differ. 2022;29(4):771–784.
22. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–723.
23. Kroemer G, Senovilla L, Galluzzi L, André F, Zitvogel L. Immunogenic cell death in cancer therapy: progress and impact on clinical outcomes. EMBO J. 2023;42(8):e113181.
Doxorubicin’in Meme Kanserinde ve Onkovirüs/Onkomantar Enfekte Konak Hücrelerde Moleküler Etki Mekanizmaları: Viral ve Fungal Replikasyon, Sinyal Yolakları, Epigenetik Düzenlemeler ve İmmün Modülasyon Bağlamında Güncel Bulgular
1. Giriş
Doxorubicin (DOX), antrasiklin grubu antineoplastik ajanlar arasında yer alan ve DNA’ya interkalasyon, topoizomeraz II inhibisyonu ile reaktif oksijen türleri (ROS) üretimi üzerinden DNA hasarı ve apoptoz indükleyen güçlü bir kemoterapötiktir [1,2].
Klasik sitotoksik etkilerinin ötesinde, son yıllarda DOX’un bağışıklık sistemini yeniden programlayıcı etkileri ön plana çıkmıştır. DOX, immünojenik hücre ölümü (ICD) oluşturmakta, tümör mikroçevresindeki baskılayıcı hücre popülasyonlarını (Treg, MDSC) azaltmakta ve antijen sunumunu artırmaktadır [3].
Özellikle meme kanseri gibi immün açıdan aktif tümörlerde, DOX’un onkovirüs (HPV, EBV) ve onkomantar (Candida spp.) enfeksiyonlarıyla etkileşimleri, bu ajanın hem antitümör hem de antimikrobiyal-immunomodülatör özelliklerini ön plana çıkarmaktadır.
2. Onkomantar Etkileşimleri
DOX’un mantar biyolojisi üzerindeki etkileri dolaylıdır. Candida albicans gibi fırsatçı patojenlerin kemoterapi sonrası konak dokularda artan kolonizasyonuna rağmen, DOX’un fungal hücre stres yanıtlarını etkileyebileceği düşünülmektedir [4].
Bazı in vitro çalışmalar, DOX’un Candida ABC taşıyıcı proteinleri (ör. CDR1, CDR2) üzerinden antifungal direnç gen ekspresyonunu modüle ettiğini ve bu yolla ilaç direnci gelişiminde rol oynayabileceğini bildirmiştir [4].
Diğer yandan, tümör dokularındaki fungal biyokütle artışı ile tümör progresyonu arasında pozitif korelasyon olduğu gösterilmiştir [5]. Bu bağlamda, DOX’un immün mikroçevrede oluşturduğu proinflamatuvar baskı ve antijenik sunum artışı, mantar-tümör etkileşim dengesini dolaylı olarak değiştirebilir.
3. Viral Replikasyon ve Onkoprotein Ekspresyonu
DOX’un viral replikasyon üzerindeki etkisi bağlama bağımlıdır.
Onkolitik adenovirüslerle birlikte uygulandığında DOX, Smad3 fosforilasyonunu artırarak viral E1A gen ekspresyonunu ve replikasyon verimliliğini yükseltir; bu da onkolitik viroterapi ile sinerjik antitümör etki yaratır [6].
Öte yandan, EBV ve HPV gibi latent onkovirüslerle enfekte hücrelerde, DOX’un oluşturduğu ICD sonucu viral antijenlerin MHC-I aracılı sunumu artar; bu da CD8⁺ T hücre aracılı antiviral bağışıklığı güçlendirir [3,5,7].
Dolayısıyla DOX, viroterapiyle sinerji ve latent viral onkoprotein ekspresyonunun baskılanması yönünde çift etkili bir ajan olarak değerlendirilmektedir.
4. Sinyal Yolakları Üzerindeki Etkiler
DOX’un çok eksenli sinyal yolağı düzenleyici etkileri bulunmaktadır:
• NF-κB baskılanması: DOX’un ROS üretimi, NF-κB’nin p65 alt birimi üzerinde inhibisyon yaparak IL-6, IL-8 ve VEGF ekspresyonunu düşürür. Bu durum inflamatuvar yanıtı ve tümör anjiyogenezini baskılar [8].
• PI3K/AKT/mTOR inhibisyonu: Bu yolak meme kanserinde kemoterapi direnci ile ilişkilidir. DOX’un PI3K/AKT/mTOR inhibitörleri (ör. piperin) ile birlikte verilmesi sinerjik apoptoz etkisi doğurur [9,10].
• MAPK (JNK/p38) aktivasyonu: DOX, DNA hasarı kaynaklı stres yanıtlarını JNK/p38 üzerinden tetikler; bu da apoptotik kaspaz kaskadını aktive eder [11].
Bu etkiler, hem tümör büyümesinin baskılanması hem de viral ve mantar enfeksiyonlarının replikatif kontrolü açısından önemlidir.
5. Epigenetik Düzenlemeler
DOX, DNA’ya interkalasyonu sonucu histon eviksiyonu ve kromatin gevşemesi yaparak epigenetik yeniden programlama etkisi oluşturur.
Bu etki hem tümör baskılayıcı genlerin yeniden ekspresyonunu, hem de entegre viral DNA bölgelerinin transkripsiyonel sessizleşmesini etkileyebilir [12].
Özellikle EBV pozitif meme kanseri modellerinde DOX’un viral LMP1 ve EBNA1 gen ekspresyonunu baskıladığı ve bu mekanizmanın DNA metilasyon artışı ile ilişkili olduğu öne sürülmektedir [3,12].
6. İmmün Kaçış Mekanizmaları
DOX’un bağışıklık kaçış mekanizmalarına müdahalesi iki yönlüdür:
1. PD-L1 mRNA destabilizasyonu: DOX, tristetraprolin (TTP) üzerinden PD-L1 mRNA stabilitesini azaltarak PD-L1 protein düzeylerini düşürür; böylece T hücre baskılanmasını hafifletir [13].
2. MHC-I artışı ve antijen sunumu: ICD aracılığıyla kalretikulin yüzey ekspresyonu ve MHC-I artışı, CD8⁺ T hücre aktivasyonunu güçlendirir [14].
Bu mekanizmalar, PD-1/PD-L1 inhibitörleriyle kombinasyon tedavilerinde sinerji potansiyeli taşır [15,16].
7. Doğuştan Gelen ve Adaptif İmmün Yanıtlar
DOX’un ICD indüksiyonu, kalretikulin (CRT) ekspresyonu, ATP ve HMGB1 salınımı gibi hasar ile ilişkili moleküler örüntülerin (DAMPs) ortaya çıkmasına neden olur [14,17].
Bu moleküller, dendritik hücreleri aktive eder, tip I interferon yanıtını tetikler ve CD8⁺ T/NK hücrelerinin tümör hedefleme kapasitesini artırır.
Sonuçta, adaptif bağışıklık aktivasyonu artarken Treg ve MDSC baskınlığı azalır.
Bu biyolojik temel, DOX’un anti-PD-1/PD-L1 ilaçlarla kombinasyonunda gözlenen klinik sinerjiyi açıklamaktadır [18].
8. Klinik Bulgular
Klinik olarak, triple-negatif meme kanseri (TNBC) hastalarında neoadjuvan DOX tedavisi sonrası tümör dokularında PD-L1 ekspresyonunun azaldığı, buna karşılık CD8⁺ T hücre infiltrasyonunun arttığı gösterilmiştir [19].
Ayrıca onkolitik virüslerle kombine erken faz çalışmalarda, DOX’un antitümör immün yanıtı artırdığı ve yanıt oranlarını yükselttiği rapor edilmiştir [6,7].
Bu veriler, DOX’un yalnızca sitotoksik değil, immün aktivatör bir ajan olarak yeniden konumlandırılabileceğini desteklemektedir.
9. Sonuç ve Mide Kanseri Bağlamında Perspektif
Doxorubicin, klasik DNA hasarı mekanizmasının ötesinde, bağışıklık sistemini yeniden şekillendiren, viral/fungal replikasyonu baskılayan, sinyal ağlarını modüle eden ve epigenetik düzeni değiştiren çok katmanlı bir ajan olarak öne çıkmaktadır.
Bizim tezimize göre, bu etkiler mide kanseri bağlamında da terapötik fayda sağlayabilir.
Özellikle EBV pozitif mide kanseri alt tiplerinde, DOX’un ICD indüksiyonu ile antijen sunumunu artırması [14,17], PD-1/PD-L1 blokajı ile sinerji oluşturması [15,16] ve viral transformasyonu baskılaması [3,12], bu ajanın immünoterapi kombinasyonlarında tamamlayıcı bir ajan olarak değerlendirilmesini desteklemektedir.
Gelecekteki faz I–II düzeyindeki klinik çalışmalar, DOX’un bu immuno-onkolojik ve mikrobiyal etkileşimsel boyutlarını aydınlatacaktır.
Kaynaklar
1. Thorn CF, Oshiro C, Marsh S, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics. 2011;21(7):440–446.
2. Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments. Pharmacol Rev. 2004;56(2):185–229.
3. Zapatka M, Borozan I, Brewer DS, et al. The landscape of viral associations in human cancers. Nat Genet. 2020;52(3):320–330.
4. Routh D, Nayak R, Mohapatra S, Dash C, Choudhury D. Anticancer drugs inhibit Candida morphogenesis. PLoS One. 2014;9(5):e98133.
5. Narunsky-Haziza L, et al. Pan-cancer mycobiome analysis reveals fungal–immune interactions. Cell. 2022;183(10):276–293.
6. Zhou X, Sun Y, Li H, et al. Doxorubicin enhances antitumor effect of oncolytic adenovirus via Smad3 phosphorylation. J Cell Mol Med. 2020;24(8):4872–4884.
7. Ranki T, Pesonen S, Hemminki A. Oncolytic adenovirus ONCOS-102 and chemotherapy synergize to induce antitumor immunity. Front Immunol. 2021;12:626050.
8. Karin M. NF-κB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1(5):a000141.
9. Zhang H, Sun X, Yang Y. PI3K/AKT/mTOR pathway in viral oncogenesis. Nat Rev Cancer. 2022;22(6):401–417.
10. Hakeem A, Khan MA, Qazi MH, et al. Piperine enhances doxorubicin cytotoxicity in breast cancer cells via PI3K/AKT/mTOR inhibition. Sci Rep. 2024;14:9955.
11. Wang J, Dong Z, Fang Y. MAPK signaling pathway regulates apoptosis via transcriptional activation. Cell Cycle. 2020;19(14):1637–1649.
12. Das PM, Singal R. DNA methylation and cancer. J Clin Oncol. 2004;22(22):4632–4642.
13. Zhai L, Bell A, Ladomersky E, et al. Tristetraprolin promotes doxorubicin-induced immunogenic cell death by destabilizing PD-L1 mRNA. Cell Death Dis. 2022;13(2):126.
14. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17(2):97–111.
15. Chen L, Gao H, Liang W, et al. Doxorubicin remodels the tumor immune microenvironment and facilitates anti-PD-1 therapy. Cancer Lett. 2024;569:216366.
16. Emens LA. Breast cancer immunotherapy: facts and hopes. Clin Cancer Res. 2018;24(3):511–520.
17. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. DAMPs, PAMPs and the origins of immunity. Immunity. 2013;39(6):1003–1018.
18. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Resistance to cancer immunotherapy. Cell. 2017;168(4):707–723.
19. Schalper KA, Carvajal-Hausdorf D, McLaughlin J, et al. Neoadjuvant chemotherapy and PD-L1 modulation in breast cancer. Ann Oncol. 2020;31(7):951–957.
20. Kroemer G, Senovilla L, Galluzzi L, André F, Zitvogel L. Immunogenic cell death and clinical outcomes. EMBO J. 2023;42(8):e113181.
Etoposide’in Meme Kanserinde ve Enfekte Konak Hücrelerde Moleküler Etkileri: Sinyal Yolakları, Epigenetik Düzenlemeler ve İmmün Modülasyon Bağlamında Güncel Değerlendirme
1. Giriş
Etoposide (VP-16), topoizomeraz II inhibitörü olarak DNA çift zincir kırıkları oluşturan ve apoptoz sürecini başlatan bir kemoterapötiktir [1]. 1980’lerden bu yana germ hücreli tümörler, akciğer kanseri, lenfomalar ve meme kanseri gibi birçok malignitede kullanılmaktadır.
Son yıllarda yapılan çalışmalar, etoposide’in yalnızca DNA kırıkları oluşturmadığını; aynı zamanda NF-κB, MAPK, PI3K/AKT/mTOR gibi hücresel sinyal yollarını etkilediğini, epigenetik yeniden programlama başlattığını ve immünojenik hücre ölümü (ICD) benzeri süreçleri indüklediğini göstermektedir [2].
Bu etkiler, özellikle viral veya fungal enfeksiyonlarla ilişkili tümör mikroçevresinde, immün cevabın yeniden şekillenmesi ve tümör immünojenitesinin artması bakımından klinik açıdan dikkat çekicidir.
2. Mantar Replikasyonu ve Onkomantar Etkileşimler
Etoposide’in fungal replikasyon üzerindeki doğrudan etkisi gösterilmemiştir. Ancak kemoterapi sonrası Candida spp. kolonizasyonu sıklıkla artmaktadır [3].
Buna rağmen bazı deneysel modellerde, β-glukan ile birlikte uygulandığında etoposide’in NF-κB aktivasyonunu baskıladığı, IL-1β ve TNF-α düzeylerini düşürdüğü ve böylece konak immün hücrelerinin duyarlılığını artırdığı rapor edilmiştir [4].
Bu bulgular, etoposide’in dolaylı olarak fungal immün yanıtı destekleyici bir rol üstlenebileceğini düşündürmektedir. Ayrıca, tümör mikroçevresinde Candida albicans antijenlerinin varlığı, tip I interferon yanıtlarını aktive ederek etoposide’in immünoterapötik etkisini güçlendirebilir [5].
3. Konak Hücre Sinyal Yolakları
3.1 NF-κB Yolu
Etoposide, DNA hasarını takiben ATM/ATR-p53 ekseni üzerinden NF-κB aktivasyonunu baskılar. Bu baskılanma, anti-apoptotik genlerin (BCL-2, XIAP) ekspresyonunu düşürür ve hücreleri apoptoza daha duyarlı hale getirir [1,6]. Ayrıca NF-κB inhibisyonu, tümör mikroçevresinde proinflamatuvar sitokin üretimini azaltarak immün baskıyı hafifletir.
3.2 MAPK Yolu
Etoposide, ERK, JNK ve p38 yolaklarını aktive ederek hücre stres yanıtını güçlendirir. Bu aktivasyon, PD-L1 ekspresyonunun azalmasına, p21-kaspaz-3 ekseninin aktive olmasına ve apoptozun hızlanmasına yol açar [7].
MAPK üzerinden oluşan fosforilasyon değişiklikleri, aynı zamanda DNA tamir proteinlerinin (RAD51, BRCA1) ekspresyonunu azaltarak kemoterapiye duyarlılığı artırır.
3.3 PI3K/AKT/mTOR Yolu
PI3K/AKT/mTOR sinyal yolunun etoposide ile baskılanması, MHC-I ekspresyonunun artmasına ve PD-L1 protein düzeylerinin azalmasına neden olur [8].
Bu etki, tümör hücrelerinin antijen sunma kapasitesini artırarak CD8⁺ T hücre aracılı immün yanıtı güçlendirir ve immün kaçışı sınırlayan önemli bir mekanizmadır.
4. Epigenetik Düzenlemeler
Etoposide’in neden olduğu DNA kırıkları, kromatin mimarisini yeniden düzenler. Özellikle histon asetilasyon ve DNA metilasyon değişiklikleri sonucunda tümör baskılayıcı genlerin (p21, p27) yeniden ekspresyonu gözlenmiştir [9].
Etoposide, HDAC inhibitörleri (ör. vorinostat, trichostatin A) ile kombine edildiğinde DNA tamir kapasitesi azalır, γ-H2AX pozitifliği artar ve hücreler immünojenik apoptoz sürecine yönelir [10].
Bu kombinasyon, hem tümör immünojenitesini artırmakta hem de ICD-benzeri antijen salınımını kolaylaştırmaktadır.
5. İmmün Kaçış Mekanizmaları
Etoposide, PI3K/AKT ve MAPK yolaklarını baskılayarak PD-L1 ekspresyonunu azaltır [7,8].
Aynı zamanda MHC-I aracılı antijen sunumunu artırarak T hücre tanıma yeteneğini güçlendirir.
Bu etkiler, anti-PD-1/PD-L1 immün kontrol noktası inhibitörleriyle kombinasyon halinde sinerjik yanıt oluşturur [11].
Bu nedenle etoposide, yalnızca kemoterapötik değil aynı zamanda immün mikroçevre düzenleyici bir ajan olarak konumlandırılabilir.
6. Doğuştan Gelen ve Adaptif İmmün Yanıtlar
Etoposide, aşırı aktive CD8⁺ T hücrelerinde seçici apoptoz oluşturarak hiperinflamasyonun önüne geçer [1].
Buna karşın, IFN-β üretimi ve IRF3/7 aktivasyonu üzerinden doğuştan gelen bağışıklığı güçlendirir [12].
Ayrıca MHC-I ekspresyonunu artırması ve DAMP salınımını tetiklemesi, dendritik hücre olgunlaşmasını hızlandırır. Bu süreçler, adaptif bağışıklığın yeniden eğitilmesine katkı sağlar
7. Klinik Bulgular
Klinik olarak, metastatik meme kanseri olgularında oral etoposide tedavisinin belirgin kısmi yanıt oranları sağladığı bildirilmiştir [13].
Ayrıca triple-negatif meme kanseri modellerinde HDAC inhibitörleriyle kombinasyon halinde uygulandığında, tümör büyümesi ve metastatik kapasite anlamlı düzeyde azalmıştır [10].
Bu bulgular, etoposide’in klasik kemoterapötik sınırlarının ötesine geçen immünomodülatör bir potansiyel taşıdığını göstermektedir.
8. Sonuç ve Mide Kanseri Bağlamında Perspektif
Etoposide, yalnızca DNA kırığı indükleyen bir ajan değil, aynı zamanda NF-κB, MAPK ve PI3K/AKT/mTOR yolaklarını düzenleyen, epigenetik yeniden programlama başlatan ve immün kaçışı sınırlayan bir immünomodülatördür.
Bu çok katmanlı etkiler, özellikle EBV pozitif mide kanseri alt tiplerinde tümör immünojenitesini artırma ve immünoterapilere duyarlılığı güçlendirme açısından dikkate değerdir.
Johnson ve arkadaşları [1], etoposide’in aktive T hücreleri selektif olarak etkilediğini göstermiştir.
Wang ve arkadaşları [8], PD-L1 regülasyonu ve MHC-I artışı üzerindeki etkisini; Liang ve arkadaşları [7] ise anti-PD-1/PD-L1 tedavileriyle sinerjisini ortaya koymuştur.
Bu veriler, gelecekte etoposide’in immünoterapilerle kombinasyon halinde, özellikle viral veya immün baskın mide kanseri alt tiplerinde, terapötik etkinliğinin araştırılmasını desteklemektedir.
Kaynaklar
1. Johnson TS, Terrell CE, Millen SH, Katz JD, Hildeman DA, Jordan MB. Etoposide selectively ablates activated T cells to control the immunoregulatory disorder hemophagocytic lymphohistiocytosis. J Immunol. 2014;192(1):84–91.
2. Li Y, Zhang H, Zhu L, et al. Combination of etoposide and HDAC inhibitor suppresses DNA repair and enhances apoptosis in breast cancer cells. Biochem Biophys Res Commun. 2023;627:1–8.
3. Pappas PG, Kauffman CA, Andes DR, et al. Clinical practice guideline for the management of candidiasis: 2016 update by the IDSA. Clin Infect Dis. 2018;66(9):1618–1621.
4. Wang H, et al. β-Glucan combined with etoposide modulates NF-κB pathway in A549 cells. Int J Mol Med. 2021;48:189.
5. Narunsky-Haziza L, et al. Pan-cancer mycobiome analysis reveals fungal–immune interactions. Cell. 2022;183(10):276–293.
6. Wang X, Shang X, Cui T, et al. Mechanisms controlling PD-L1 expression in cancer. Cancer Lett. 2020;468:27–38.
7. Liang SC, Luan JB, et al. Strategies targeting PD-L1 expression and associated opportunities. Int J Mol Sci. 2023;24:7890.
8. Wang X, Shang X, Cui T, et al. PI3K/AKT/mTOR regulation of PD-L1 and MHC-I in cancer immunity. Cancer Lett. 2020;468:27–38.
9. Das PM, Singal R. DNA methylation and cancer. J Clin Oncol. 2004;22(22):4632–4642.
10. Li Y, Zhang H, Zhu L, et al. HDAC inhibition potentiates etoposide-induced apoptosis. Biochem Biophys Res Commun. 2023;627:1–8.
11. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Resistance to cancer immunotherapy. Cell. 2017;168(4):707–723.
12. Struzik J, Szulc-Dąbrowska L. NF-κB signaling in oncolytic virus therapy and immune modulation. Biomed Res Int. 2018;2018:1–13.
13. Smith IE, Wardley AM, et al. Long-term oral etoposide in metastatic breast cancer. Eur J Cancer Clin Oncol. 1993;29:1107–1112.
Flutamide’in Meme Kanserinde Moleküler Etkileri ve Onkoviral/Onkomantar Etkileşimleri: Güncel Bulgular ve Mide Kanseri Perspektifi
1. Giriş
Androjen reseptör (AR) sinyal yolları, özellikle östrojen reseptörü negatif (ER–) ve HER2 pozitif meme kanseri alt tiplerinde, hücre proliferasyonu ve gen ekspresyon kontrolünde kritik rol oynar [1].
Flutamide, non-steroidal bir AR antagonisti olup uzun yıllardır prostat kanseri tedavisinde kullanılmaktadır. Son yıllarda yapılan moleküler analizler, AR pozitif (moleküler apokrin) meme kanserlerinde Flutamide’in antiproliferatif, DNA onarım baskılayıcı ve immün mikroçevre düzenleyici etkiler gösterebileceğini ortaya koymuştur [2,3].
Bu kapsamda, Flutamide’in yalnızca hormonal blokaj sağlamadığı; aynı zamanda MAPK/ERK, PI3K/AKT/mTOR ve NF-κB gibi sinyal ağlarını etkileyerek tümör hücrelerinin biyolojik davranışını yeniden şekillendirdiği anlaşılmaktadır [1,4].
2. Moleküler Mekanizmalar
2.1 Proliferasyon ve Apoptoz
AR aktivasyonu, Cyclin D1, CDK4/6 ve E2F1 gibi hücre döngüsü proteinlerini aktive eder.
Flutamide, bu ekseni bloke ederek G1 fazı duraksaması ve apoptoz indükler [2].
Ayrıca MEK inhibitörleri (ör. CI-1040) ile birlikte kullanıldığında p-ERK1/2 fosforilasyonunu azaltır ve kasapaz-3 aktivasyonu yoluyla sinerjik apoptoz etkisi oluşturur [2,3].
2.2 AR–MEK/ERK ve PI3K/mTOR Ekseni
AR sinyali, MEK/ERK ve PI3K/AKT/mTOR yolaklarıyla kesişir. Bu yolaklar, hem hücre proliferasyonunu hem de anti-apoptotik sinyalleri destekler.
Flutamide, ERK1/2 fosforilasyonunu ve AKT/mTOR aktivitesini baskılayarak hücre büyümesini durdurur [2,4].
Bu durum, AR blokajının yalnızca transkripsiyonel değil aynı zamanda sinyal ağ düzeyinde antiproliferatif bir etki yarattığını gösterir.
2.3 DNA Hasar Onarımı ve Genomik Stabilite
AR, DNA tamirinde özellikle non-homologous end joining (NHEJ) ve BRCA1/2 bağımlı onarım süreçlerinde görev alır.
Flutamide tedavisi, DNA onarım proteinlerini (KU70, XRCC4, RAD51) azaltarak DNA hasarına duyarlılığı artırır [1,5].
Bu etki, γ-H2AX birikimi ve apoptoz oranlarında artış ile koreledir. Özellikle DNA tamir yetersizliğinin bulunduğu tümörlerde bu mekanizma Flutamide’in etkinliğini artırabilir.
3. Onkovirüs ve Onkomantar Etkileşimleri
3.1 Onkovirüslerle Etkileşim
Epstein–Barr virüsü (EBV) ve human papillomavirüs (HPV) gibi onkovirüsler, NF-κB ve STAT3 yolaklarını aktive ederek viral onkoprotein ekspresyonunu artırır [6].
Bu aktivasyon aynı zamanda AR sinyaliyle pozitif geri besleme oluşturabilir.
Flutamide, AR inhibisyonu yoluyla NF-κB/STAT3 aktivitesini baskılayarak viral gen ekspresyonunu dolaylı biçimde azaltır [7].
Bu etki, özellikle EBV pozitif meme ve mide kanseri alt tiplerinde viral latent proteinlerin (LMP1, EBNA1) ekspresyonunu sınırlayarak immünoterapötik duyarlılığı artırabilir [6–8].
3.2 Onkomantar Etkileşimleri
Candida spp. gibi fırsatçı mantarlar, NF-κB ve ERK yolaklarını aktive ederek kronik inflamasyon ve tümör destekleyici sitokin salınımı oluşturur [9].
AR aktivitesi bu inflamatuvar sinyallerle etkileşebilir.
Flutamide tedavisi, NF-κB baskılanması ve IL-6/IL-8 ekspresyonunun azalması yoluyla onkomantar kaynaklı inflamatuvar mikroçevreyi zayıflatabilir.
Bu dolaylı etki, özellikle immün baskı altındaki hastalarda fungal patogenezle ilişkili tümör progresyonunu sınırlandırma potansiyeli taşır [9,10].
4. Moleküler Belirteçler
Flutamide’in biyolojik etkinliği çoklu belirteçlerle izlenebilir:
• AR çekirdek lokalizasyonu (IHK): Tedavi sonrası AR nükleer translokasyonu azalır [1].
• p-ERK1/2 düzeyleri: MEK/ERK aktivasyonundaki baskıyı yansıtır [2].
• Ki-67 indeksi: Proliferasyon oranını gösterir; Flutamide ile azalır [3].
• γ-H2AX birikimi: DNA hasarı düzeyini yansıtır; Flutamide sonrası artış gözlenir [1].
• PD-L1 ekspresyonu: İmmün mikroçevredeki dönüşümün göstergesidir; Flutamide tedavisiyle azalır [7,8].
Bu belirteçler, Flutamide’in hem proliferatif hem immünomodülatör etkilerini ölçmede klinik açıdan değerlidir.
5. Klinik Bulgular
Flutamide, Faz II klinik çalışmalarda tek ajan olarak sınırlı etkinlik göstermiştir [4].
Ancak MEK inhibitörleri (ör. CI-1040) veya HDAC inhibitörleri ile kombinasyon, AR pozitif moleküler apokrin meme kanseri modellerinde tümör büyümesini anlamlı biçimde azaltmıştır [2].
Ayrıca, AR pozitif ve viral yükü yüksek tümörlerde, Flutamide’in PD-L1 ekspresyonunu azaltıcı ve T hücre infiltrasyonunu artırıcı etkileri gözlemlenmiştir [6–8].
Bu bulgular, Flutamide’in immün-onkolojik açıdan da değerlendirilebileceğini göstermektedir.
6. Klinik Yaklaşımlar ve Kombinasyon Stratejileri
Flutamide’in etkinliğini artırmak için önerilen kombinasyon yaklaşımları şunlardır:
1. MEK inhibitörleri (CI-1040, Trametinib) → Sinyal baskılanması ve apoptoz sinerjisi [2].
2. DNA tamir baskılayıcı ajanlar (PARP inhibitörleri) → BRCA1/2 eksikliğinde etkinlik artışı [5].
3. PD-1/PD-L1 blokajı ile kombinasyon → İmmün mikroçevre yeniden programlanması [7,8].
Bu kombinasyonlar, hem proliferatif hem immün kaçış mekanizmalarını hedefleyen rasyonel çoklu tedavi stratejileri sunmaktadır.
7. Mide Kanseri Bağlamında Perspektif
Androjen reseptör sinyali, özellikle EBV pozitif mide kanseri alt tiplerinde aktif rol oynayabilir [6,8].
Flutamide’in DNA tamir baskısı, PD-L1 azalması, T hücresi aktivasyonu ve NF-κB/STAT3 baskılanması yoluyla EBV pozitif mide kanserlerinde terapötik potansiyel taşıdığı düşünülmektedir.
AR–viral sinyal kesişimi, bu tümörlerde immünoterapötik yaklaşımlarla kombine edilerek faz I/II klinik çalışmalar düzeyinde değerlendirilmelidir [8,11].
8. Tartışma ve Sonuç
Flutamide, klasik androjen reseptör antagonizmasının ötesine geçen, sinyal yolakları, DNA onarımı, epigenetik kontrol ve immün mikroçevre üzerinde çok eksenli etkiler sergileyen bir ajan olarak yeniden değerlendirilmelidir.
Özellikle MEK/ERK eksenindeki baskı, proliferatif sinyallerin susturulması açısından önemli bir kazanımdır [2,4].
Ayrıca, AR sinyalinin EBV ve HPV kaynaklı onkoviral proteinlerle etkileşimi, Flutamide’in virüs ilişkili meme ve mide kanserlerinde potansiyel kullanımını desteklemektedir [6–8].
Gelecekte yapılacak klinik çalışmalarda, AR ekspresyonu yüksek, PD-L1 pozitif veya viral yüklü tümörlerde Flutamide’in MEK inhibitörleri ve immün kontrol noktası blokajı ile birlikte test edilmesi önerilmektedir.
Kaynaklar
1. Treré D, La Monica S, et al. Understanding androgen receptor signaling in non-prostatic malignancies. NPJ Breast Cancer. 2020;6:86.
2. Chia KM, et al. Flutamide synergistically enhances MEK inhibitor CI-1040 in molecular apocrine breast cancer. Breast Cancer Res. 2010;12(3):R41.
3. Di Monaco M, et al. Inhibitory effect of hydroxyflutamide plus tamoxifen in MCF-7 cells. J Cancer Res Clin Oncol. 1995;121(12):710–714.
4. Perrault DJ, et al. Phase II study of flutamide in metastatic breast cancer. Eur J Cancer. 1988;24(3):319–323.
5. Hsu CL, Chen Y, et al. Androgen receptor regulates DNA repair and radiosensitivity in cancer. Mol Cancer Res. 2023;21(4):501–512.
6. Moinfar F, et al. Androgen receptors frequently expressed in breast carcinoma. Cancer. 2003;98(4):703–711.
7. Xu Y, et al. Androgen signaling modulates tumor immune microenvironment in EBV-associated cancers. Front Immunol. 2024;15:40504439.
8. Bhatia-Gaur R, et al. Androgen receptor-mediated signaling in gastric cancer. Cancers (Basel). 2024;16(18):3219.
9. Conti HR, Gaffen SL. Candida albicans and inflammation: the host perspective. Pathog Dis. 2015;73:ftv075.
10. Narunsky-Haziza L, et al. Pan-cancer mycobiome analysis reveals fungal–immune interactions. Cell. 2022;183(10):276–293.
11. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–723.
12. Kroemer G, Senovilla L, Galluzzi L, André F, Zitvogel L. Immunogenic cell death and clinical outcomes. EMBO J. 2023;42(8):e113181.
Mitobronitol’un Meme Kanseri ve Onkovirüs/Onkomantar Etkileşimlerindeki Moleküler Etkileri: Mide Kanseri Bağlamında Perspektif
Özet
Mitobronitol, brom içeren bir mannitol türevi olup DNA alkilleyici ajanlar sınıfında yer alır. Klasik DNA hasarı ve apoptoz mekanizmalarının ötesinde, son dönemde yapılan teorik ve preklinik analizler bu molekülün mitokondriyal enerji metabolizması, epigenetik yanıtlar ve bağışıklık mikroçevresi üzerinde düzenleyici etkiler oluşturabileceğini düşündürmektedir. Bu derleme, Mitobronitol’un meme kanserindeki olası moleküler etki mekanizmalarını, onkovirüs ve onkomantar etkileşimleriyle ilişkisini ve EBV pozitif mide kanseri bağlamındaki potansiyel terapötik yansımalarını ele almaktadır.
1. Giriş
Meme kanseri, heterojen moleküler alt tipleri ve immün mikroçevresindeki karmaşık etkileşimler nedeniyle çok hedefli yaklaşımlar gerektirir [1].
Onkovirüsler (ör. Epstein–Barr virüsü [EBV], human papillomavirus [HPV]) ve onkomantarlar (ör. Candida albicans, Malassezia spp.), NF-κB, STAT3, TGF-β ve epigenetik yeniden programlama eksenleri üzerinden kronik inflamasyon ve tümör progresyonuna katkıda bulunurlar [1–3].
Mitobronitol, bromlu yapısı nedeniyle lipofilik özellik gösteren ve hem DNA hem de mitokondriyal fonksiyonlar üzerinde etkili olabileceği öne sürülen bir alkilleyici ajandır [4]. Bu yönüyle, patojen kaynaklı sinyal ağlarının modülasyonu ve immün mikroçevrenin yeniden programlanması açısından incelenmeye değerdir.
2. Mitobronitol’un Moleküler Etki Mekanizmaları
2.1 DNA Hasarı ve Hücre Döngüsü Kontrolü
Mitobronitol, DNA’da interstrand cross-link oluşturarak replikasyonu durdurur ve çift zincir kırıkları meydana getirir. Bu durum p53 bağımlı veya p53 bağımsız apoptoz süreçlerini aktive eder [4].
Hasar yanıtı sonucunda G2/M fazında hücre döngüsü duraklar; CHK1/CHK2 aktivasyonu ile mitotik ölüm (mitotic catastrophe) gözlenebilir.
Bu mekanizma, γ-H2AX birikimi ve cleaved caspase-3 ekspresyonu ile doğrulanabilir [5].
2.2 Mitokondriyal Metabolizma ve Oksidatif Stres
Mitobronitol’un bromlu yapısı, membran geçirgenliğini artırarak mitokondriyal membran potansiyelinde bozulma, ROS artışı ve ATP düşüşü ile sonuçlanabilir.
Buna karşılık AMPK aktivasyonu ve mTOR baskılanması, hücrede enerji stres yanıtını tetikler [5,6].
Bu süreç, autofaji-apoptoz dengesi ve immünojenik hücre ölümü (ICD) açısından önem taşır.
2.3 Sinyal Yolaklarının Düzenlenmesi
Mitobronitol, DNA hasarına ikincil olarak NF-κB, STAT3 ve PI3K/AKT/mTOR sinyallerini baskılayabilir.
Benzer etkiler, Ciclopirox ve Cytarabine gibi DNA hasarı oluşturan ajanlarda gösterilmiştir [5,7,8].
Bu sinyallerin baskılanması, PD-L1 ekspresyonunun azalmasına ve T hücresi aktivasyonunun artmasına katkı sağlayabilir [9].
3. Onkovirüs ve Onkomantar Etkileşimleri
3.1 Onkovirüsler
HPV’nin E6/E7, EBV’nin LMP1/EBNA1 onkoproteinleri p53 ve Rb yolaklarını baskılar, bu da hücresel onarım mekanizmalarını zayıflatır [10].
Mitobronitol’un indüklediği DNA hasar yanıtı, bu viral proteinlerin etkilerini dolaylı biçimde zayıflatabilir; ayrıca kromatin yeniden düzenlenmesi yoluyla viral gen ekspresyonunu baskılayabilir.
EBV-pozitif hücrelerde DNA hasarı sonrası viral replikasyonun baskılandığı ve LMP1 düzeylerinin azaldığı benzer ajanlarla gösterilmiştir [6,10]. Bu etki, viral immün kaçışın kırılmasına katkı sunabilir.
3.2 Onkomantarlar
Candida albicans ve Malassezia spp. gibi mantarlar, NF-κB/STAT3 aktivasyonu yoluyla kronik inflamatuvar mikroçevre oluşturur [3,7].
Mitobronitol’un doğrudan antifungal etkisi kanıtlanmamış olsa da, DNA hasarına bağlı proliferatif sinyal azalması ve inflamatuvar sitokin baskılanması dolaylı olarak mantar kaynaklı tümör destekleyici ortamı zayıflatabilir [3,9].
Ayrıca fungal β-glukan uyarımıyla tetiklenen Toll-like reseptör (TLR2/4) sinyalleri, Mitobronitol’un NF-κB modülasyonu ile sinerjik biçimde baskılanabilir [3,9].
4. Mitobronitol’un Diğer Alkilleyici Ajanlarla Karşılaştırmalı Etkileri
Mitobronitol, bromlu mannitol çekirdeği nedeniyle Cytarabine veya Busulfan gibi ajanlardan farklı lipofilik özellikler gösterir.
Bu farklılık, hücre içi dağılımı, mitokondriyal etkileşimi ve enerji metabolizması üzerindeki etkilerini değiştirebilir [4,8].
Benzer ajanlarda gözlenen γ-H2AX birikimi, p53 aktivasyonu ve G2/M faz duraklaması, Mitobronitol için öngörülen moleküler imzayı oluşturur [5,8].
Ayrıca Cytarabine’in PD-L1 ekspresyonunu azalttığı ve immün kontrol noktası inhibitörleriyle sinerji sağladığı bilinmektedir; Mitobronitol’un da benzer DNA hasar-bağışıklık aktivasyonu etkileri gösterebileceği düşünülmektedir [9].
5. Klinik ve Deneysel Perspektif
Mitobronitol’un klinik kullanım verisi bulunmamaktadır; mevcut bilgiler yalnızca preklinik ve teorik düzeyde sınırlıdır [4].
Gelecekteki çalışmaların aşağıdaki biyobelirteçlere odaklanması önerilir:
• Ki-67 → Hücre proliferasyonu
• γ-H2AX → DNA çift zincir kırıkları
• Cleaved caspase-3 → Apoptoz
• p-STAT3 → İnflamasyon aktivitesi
• PD-L1 → İmmün kaçış durumu
• CD8⁺ T hücre infiltrasyonu → Antitümör bağışıklık
• EBV/HPV viral yükü ve fungal ITS1/ITS2 dizileri → Mikrobiyal katkı değerlendirmesi
Bu parametreler, Mitobronitol’un immün ve metabolik etkilerini deneysel olarak doğrulamak açısından önemlidir.
6. Tartışma
Mitobronitol’un etkileri yalnızca DNA alkilleme ve sitotoksisite ile sınırlı değildir.
Mitokondriyal stres, AMPK/mTOR aksında düzenleme, ROS aracılı sinyal modülasyonu ve NF-κB/STAT3 baskısı, ajanı metabolik-immünolojik yeniden programlayıcı bir molekül haline getirmektedir [5,6].
Bu süreçler, immün baskılayıcı hücre popülasyonlarının (Treg, MDSC) azalmasına, PD-L1 regülasyonunun değişmesine ve T hücre infiltrasyonunun artmasına yol açabilir.
Özellikle EBV pozitif mide kanserlerinde, DNA hasar yanıtı ile viral onkoproteinlerin baskılanması, PD-1/PD-L1 blokajı ile sinerjik antitümör etki yaratma potansiyeli taşır [9–11].
Ancak bu hipotezlerin doğrulanması için, hem in vitro genotoksisite modelleri hem de in vivo immün yanıt analizleri gereklidir.
7. Sonuç
Mitobronitol, klasik bir DNA alkilleyici ajan olmasının ötesinde, mitokondriyal stres, sinyal yolu modülasyonu ve immün mikroçevre yeniden programlaması gibi mekanizmalarla potansiyel olarak immünoterapötik etkiyi destekleyen bir bileşik olarak değerlendirilmektedir.
Özellikle EBV pozitif, PD-L1 yüksek ekspresyonlu mide kanseri alt tiplerinde, immün kontrol noktası inhibitörleri ile kombinasyon halinde faz I/II düzeyinde test edilmesi teorik olarak anlamlıdır.
Bu bağlamda, Mitobronitol gelecekte DNA hasar-immün aktivasyon sinerjisi odaklı yeni nesil tedavi stratejilerinde yer alma potansiyeline sahiptir.
Kaynaklar
1. Gullo F, et al. Microbiome-fungal interactions in breast cancer. Cell Mol Life Sci. 2021;78(3):927-940.
2. Rajagopala SV, et al. The human microbiome and cancer. Nat Rev Cancer. 2020;20(11):745-762.
3. Gaffen SL, et al. Fungal pathogenesis and cancer progression via immune modulation. Pathog Dis. 2016;74(5):ftw019.
4. PubChem Database. Mitobronitol compound summary. [Accessed 2025].
5. Feng Z, et al. Ciclopirox targets p-STAT3 and AMPK-mTOR in cancer cells. Oncogene. 2021;40:551-563.
6. Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10(8):550-560.
7. Pfaller MA, Diekema DJ. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev. 2007;20(1):133-163.
8. Assaraf YG, et al. Molecular basis of resistance to cytarabine in cancer therapy. Drug Resist Updat. 2020;50:100695.
9. Song Y, et al. Cytarabine sensitizes breast cancer cells to immune checkpoint blockade via DNA damage response. J Exp Clin Cancer Res. 2022;41:164.
10. Narunsky-Haziza L, et al. Pan-cancer mycobiome analysis reveals fungal-immune interactions. Cell. 2022;183(10):276-293.
11. Shinozaki-Ushiku A, Kunita A, Fukayama M. Update on Epstein–Barr virus and gastric cancer. Gastric Cancer. 2015;18(1):12-22.
12. Kroemer G, Senovilla L, Galluzzi L, André F, Zitvogel L. Immunogenic cell death and clinical outcomes. EMBO J. 2023;42(8):e113181.
Thiram’ın Meme Kanseri, Onkovirüs ve Onkomantar Enfekte Konak Hücrelerde Moleküler Etki Mekanizmaları: Sinyal Yolakları, Epigenetik Modifikasyonlar ve İmmün Kaçış Bağlamında Güncel Bulgular
Özet
Thiram (tetrametiltiyuram disülfid), ditiokarbamat sınıfına ait yaygın kullanılan bir fungisittir. Klasik pestisit etkilerinin ötesinde, son yıllarda yapılan preklinik çalışmalar Thiram’ın anti-anjiyogenik, Wnt/β-catenin baskılayıcı, pro-oksidan ve immün modülatör özellikler gösterebileceğini ortaya koymuştur. Bu derleme, Thiram’ın meme kanserinde, onkovirüs/onkomantar ilişkili tümör mikroçevresinde ve EBV pozitif mide kanseri bağlamında potansiyel moleküler, epigenetik ve immünolojik etkilerini güncel literatür ışığında tartışmaktadır.
1. Giriş
Meme kanseri, genetik, epigenetik ve mikrobiyal faktörlerin etkileşimiyle şekillenen, yüksek heterojeniteye sahip bir malignitedir [1,2].
Onkovirüsler (ör. Epstein–Barr virüsü [EBV], human papillomavirus [HPV]) ve onkomantarlar (Candida spp., Malassezia spp.) tümör mikroçevresinde NF-κB, STAT3 ve Wnt/β-catenin sinyal yollarını aktive ederek inflamasyon, immün kaçış ve proliferasyonu destekler [3,4].
Thiram, tarımsal alanda fungisit olarak kullanılan bir bileşik olmasına karşın, Cu/Zn-SOD inhibisyonu ve Wnt baskısı yoluyla anti-tümör etkiler oluşturabileceği gösterilmiştir [5,6]. Bu etkiler, özellikle mikrobiyal sinyalle ilişkili tümörlerde translasyonel açıdan dikkat çekicidir.
2. Tümör Büyümesi ve Anjiyogenez
Thiram, C6 glioma ve Lewis akciğer karsinom modellerinde oral uygulama sonrası tümör hacminde %50–60 azalma ve metastatik nodül sayısında 3–5 kat düşüş sağlamıştır [7].
Bu etki, Cu/Zn-SOD inhibisyonu ile reaktif oksijen türlerinin (ROS) birikmesi sonucu VEGF baskısı ve anjiyogenez azalması ile ilişkilidir [8].
ROS artışı aynı zamanda endotelyal hücre proliferasyonunun azalmasına, HIF-1α ekspresyonunun düşmesine ve vasküler regresyona yol açar.
Bu mekanizma, yüksek mikrovasküler yoğunluğa sahip meme kanseri alt tiplerinde (örn. triple negatif) teorik terapötik avantaj sağlayabilir.
3. Wnt/β-Catenin ve 11β-HSD2 İnhibisyonu
Thiram, 11β-hidroksisteroid dehidrogenaz tip 2 (11β-HSD2) enzimini inhibe ederek glukokortikoid metabolizmasını ve β-catenin stabilitesini düzenler [6,9].
Bu durum, MMP-7, Cyclin D1 ve c-MYC gibi Wnt hedef genlerinin ekspresyonunun azalmasına yol açar [9].
Wnt baskılanması, aynı zamanda CD8⁺ T hücre infiltrasyonunu artırarak ve PD-L1 ekspresyonunu azaltarak immün kaçışın sınırlanmasına katkı sağlar [10].
Dolayısıyla Thiram, Wnt–mTOR ekseninin baskılanmasıyla hem proliferatif hem de immünolojik yeniden programlama etkileri gösterebilir.
4. Sinyal Yolakları ve Hücresel Etkileşimler
Thiram’ın NF-κB, MAPK ve PI3K/AKT/mTOR yolakları üzerindeki doğrudan etkileri sınırlı rapor edilmiştir.
Ancak Wnt/β-catenin ve ERK/mTOR eksenindeki baskısı, inflamasyonu, hücre hayatta kalımını ve proliferasyonu azaltabilir [6,11].
Ayrıca ROS artışı, AMPK aktivasyonu ve mTOR baskısı ile enerji stres yanıtı tetiklenir.
Bu süreç, tümör mikroçevresinde MDSC ve Treg hücrelerinin azalmasıyla anti-tümör immüniteyi güçlendirebilir.
Bu mekanizmalar, viral/mantar kaynaklı inflamatuvar sinyallerle kesişerek, Thiram’ın dolaylı immün modülatör etkilerini destekler.
5. Epigenetik Modifikasyonlar
Thiram’ın DNA metilasyonu ve histon modifikasyonları üzerindeki doğrudan etkisi bilinmemektedir.
Ancak Wnt/β-catenin baskısı, TET (Ten-eleven translocation) enzim aktivitesi, DNMT1 ekspresyonu ve histon asetiltransferaz (HAT) aktivitesini dolaylı olarak etkileyebilir [12].
Bu durum, tümör baskılayıcı genlerin (p21, APC, DKK1) yeniden ekspresyonuna ve immünojenik fenotipin güçlenmesine zemin hazırlar.
Dolayısıyla Thiram, epigenetik yeniden programlama yoluyla tümör immünojenitesini artırma potansiyeli taşır.
6. Onkovirüs ve Onkomantar Etkileşimleri
6.1 Onkovirüsler
EBV ve HPV, viral onkoproteinleri (ör. LMP1, E6/E7) aracılığıyla Wnt/β-catenin, NF-κB ve STAT3 yolaklarını aktive eder [13,14].
Thiram’ın Wnt baskısı, bu viral onkoproteinlerin transkripsiyonel etkilerini dolaylı biçimde azaltabilir.
Ayrıca, Wnt-NF-κB etkileşiminin zayıflatılması, viral reaktivasyonun baskılanmasına katkı sağlayabilir.
Bu mekanizma, özellikle EBV pozitif mide kanserlerinde translasyonel açıdan önemli bir hedef sunar [14].
6.2 Onkomantarlar
Candida albicans ve Malassezia spp., STAT3 ve NF-κB aktivasyonu yoluyla IL-6, TNF-α ve IL-23 üretimini artırır [3,15].
Bu inflamatuvar yanıt, tümör mikroçevresinde immün baskı ve anjiyogenez artışı ile sonuçlanır.
Thiram’ın dolaylı olarak NF-κB/STAT3 baskısı ve ROS artışı yoluyla bu mikroçevreyi zayıflatabileceği öne sürülmektedir.
Ayrıca antifungal kökenli olması nedeniyle Candida biyofilmlerinin gelişimini sınırlayabileceği düşünülmektedir — bu da onkomantar kaynaklı inflamasyonun azaltılmasına katkı sağlayabilir.
7. İmmün Kaçış ve Bağışıklık Yanıtı
Wnt baskısı, PD-L1 ekspresyonunun azalmasına ve T hücre infiltrasyonunun artmasına katkı sağlar [10].
Ancak Thiram’ın NK hücre fonksiyonlarını baskıladığı ve perforin/granzim-B üretimini azalttığı rapor edilmiştir [16].
Bu durum, viral enfeksiyon kontrolü ve tümör immün gözetimi açısından potansiyel bir dezavantajdır.
Dolayısıyla Thiram, immün mikroçevrede çift yönlü etki gösterir: bir yandan Wnt aracılı immün kaçışı baskılarken, diğer yandan doğuştan gelen immün yanıtı zayıflatabilir.
8. Klinik Bulgular ve Translaksyonel Yaklaşım
Thiram’ın insanlarda doğrudan kanser tedavisinde klinik kullanımı bulunmamaktadır.
Ancak aynı sınıftan ditiokarbamat türevleri (ör. disülfiram), klinik çalışmalarda tümör baskılayıcı etkiler göstermiştir [17].
Bu benzerlik, Thiram’ın da translasyonel aday molekül olarak araştırılabileceğini düşündürmektedir.
Kombine yaklaşımlar — özellikle anti-PD-1/PD-L1 veya anti-VEGF tedavileriyle — Thiram’ın anti-anjiyogenik ve Wnt baskılayıcı etkilerini sinerjik biçimde güçlendirebilir.
9. Tartışma
Thiram, klasik bir fungisit olmasına rağmen, çok eksenli tümör modülasyonu sağlayan bir bileşik olarak dikkat çekmektedir.
Anti-anjiyogenik etkisi, Wnt baskısı, epigenetik yeniden programlama potansiyeli ve viral/mantar ilişkili inflamasyonun sınırlandırılması, onu kanser immün metabolizmasının yeniden düzenlenmesinde teorik olarak değerli kılar.
Ancak NK hücre baskısı ve olası toksisite riskleri, klinik ilerleme açısından önemli kısıtlayıcı faktörlerdir [16].
10. Sonuç ve Mide Kanseri Perspektifi
Thiram’ın Wnt/β-catenin baskılayıcı ve anti-anjiyogenik özellikleri, özellikle EBV pozitif, yüksek Wnt aktivasyonlu mide kanseri alt tiplerinde teorik terapötik fayda sağlayabilir.
Ancak NK hücre fonksiyonlarını zayıflatma riski, bu ajanın yalnızca kombine tedavi stratejilerinde ve yakın immün monitörizasyon altında kullanılabileceğini düşündürmektedir.
Gelecekte, Thiram’ın DNA hasarı, ROS dengesi ve epigenetik yeniden programlama kombinasyonuyla mikrobiyal sinyal modülasyonunu hedefleyen hibrit immünoterapilerde değerlendirilmesi önerilmektedir.
Kaynaklar
1. Gullo F, et al. Microbiome-fungal interactions in breast cancer. Cell Mol Life Sci. 2021;78(3):927–940.
2. Rajagopala SV, et al. The human microbiome and cancer. Nat Rev Cancer. 2020;20:745–762.
3. Gaffen SL, et al. Fungal pathogenesis and cancer progression via immune modulation. Pathog Dis. 2016;74(5):ftw019.
4. McIlwain DR, et al. Cancer immunology and immune evasion. Nat Rev Cancer. 2023;23:75–91.
5. Li Z, et al. Wnt/β-catenin signaling in cancer: therapeutic targeting strategies. Front Pharmacol. 2021;12:762402.
6. Chang J, Xue M, et al. 11β-HSD2 inhibition suppresses lung carcinogenesis via ERK/mTOR blockade. PLoS One. 2015;10:e0127030.
7. Kochetkov N, et al. Thiram inhibits glioma growth via SOD inhibition and anti-angiogenesis. Biochemistry (Moscow). 2002;67(2):210–216.
8. Zhang Y, et al. Anti-angiogenic therapies in cancer: from bench to bedside. Signal Transduct Target Ther. 2022;7:263.
9. Wang Z, et al. Targeting β-catenin signaling for cancer therapy. Cancers (Basel). 2022;14:321.
10. Luke JJ, et al. Targeting Wnt/β-catenin pathway to enhance cancer immunotherapy. Nat Rev Clin Oncol. 2019;16:121–130.
11. Sun Y, et al. Crosstalk between Wnt/β-catenin and PI3K/AKT/mTOR signaling in cancer. Cancer Lett. 2022;531:1–12.
12. Song Y, et al. Epigenetic regulation in Wnt signaling pathway. Mol Cancer. 2020;19:184.
13. Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10(8):550–560.
14. Shinozaki-Ushiku A, et al. EBV-associated gastric cancer: focus on immune response and molecular biology. Int J Mol Sci. 2021;22:12184.
15. Pfaller MA, Diekema DJ. Epidemiology of invasive candidiasis. Clin Microbiol Rev. 2007;20(1):133–163.
16. Ma J, et al. Thiram suppresses perforin and granzyme B production in human NK cells. Immunopharmacol Immunotoxicol. 2015;37(1):72–79.
17. Skrott Z, et al. Disulfiram’s antineoplastic activity: mechanisms and clinical applications. Nat Commun. 2017;8:13957.
Tribenoside’in Moleküler Etki Mekanizmaları: İmmün, Enflamatuar ve Rejeneratif Sistemler Bağlamında Güncel Bir Derleme
Özet
Tribenoside, yarı sentetik bir glikosid olup uzun süredir hemoroid ve kronik venöz yetmezlik gibi vasküler bozukluklarda kullanılan bir venotropik ve anti-inflamatuvar ajandır [1,2]. Son yıllarda yapılan moleküler biyoloji, doku mühendisliği ve translasyonel tıp çalışmaları, tribenoside’in yalnızca vasküler tonusu düzenleyen bir bileşik olmadığını; aynı zamanda fibroblast aktivasyonu, ekstrasellüler matriks (ECM) yeniden yapılanması, mast hücre stabilizasyonu ve oksidatif stres kontrolü yoluyla rejeneratif ve immünmodülatör etkiler sergilediğini göstermektedir [3–5].
Bu derleme, 2018–2025 dönemi literatürü ışığında, tribenoside’in moleküler etki yollarını, immün-inflamatuvar sistemler üzerindeki düzenleyici rollerini ve onkolojik mikroçevre bağlamındaki olası translasyonel potansiyelini kapsamlı biçimde değerlendirmektedir.
1. Giriş
Tribenoside, vasküler permeabiliteyi azaltan, kapiller bütünlüğü koruyan, lenfatik drenajı artıran ve lokal inflamasyonu modüle eden bir ajandır [1–4].
İlk olarak venöz tonus artırıcı olarak tanımlansa da, daha yeni çalışmalar bu molekülün mast hücre dengelemesi, fibroblast migrasyonu, laminin-332 ekspresyonu ve oksidatif stres azaltımı gibi rejeneratif süreçleri destekleyebileceğini göstermektedir [5–7].
Bu etkiler yalnızca damar duvarında değil, tümör mikroçevresi, doku onarımı ve immün homeostaz açısından da ilgi çekicidir [8].
2. Moleküler Mekanizmalar
2.1 İnflamatuvar Yanıtın Modülasyonu
Tribenoside, mast hücrelerinden histamin, triptaz ve prostaglandin E₂ salınımını azaltarak inflamatuvar yanıtı sınırlandırır [5,7].
Bu etki, NF-κB ve p38 MAPK aktivitesinin baskılanmasıyla ilişkilidir.
Ayrıca TNF-α, IL-1β ve IL-6 üretimini azaltarak kronik inflamasyonun tümör progresyonuna katkısını dolaylı biçimde engelleyebilir [8].
Bu mekanizma, özellikle EBV pozitif mide kanseri veya inflamasyon baskın kolorektal tümörler gibi mikroçevre duyarlı malignitelerde önem taşır.
2.2 Kapiller Geçirgenlik ve Mikrosirkülasyon
Tribenoside, endotelyal hücre adezyonunu güçlendirerek ve VEGF kaynaklı vasküler sızıntıyı baskılayarak mikrosirkülasyonu iyileştirir [9].
Bu durum hipoksi-indüklenebilir faktör (HIF-1α) aktivitesini azaltır, anjiyogenez ve stromal yeniden yapılanma süreçlerini dengeler.
Dolayısıyla, HIF-1α–VEGF ekseninin dolaylı baskılanmasıyla hipoksiye bağlı tümör ilerlemesi sınırlandırılabilir [6,9].
2.3 Fibroblast Aktivasyonu ve ECM Yeniden Yapılanması
In vitro modellerde tribenoside’in, laminin-332 ve laminin-α5 ekspresyonunu artırarak fibroblast migrasyonunu ve bazal membran bütünlüğünü desteklediği gösterilmiştir [5,10].
ECM yeniden yapılanması, tümör invazyonunun fiziksel sınırlandırılması ve doku stabilizasyonunun korunması açısından önemli bir mekanizmadır.
Bu etki, MMP-2/9 dengesinin korunması ve integrin-fokal adezyon komplekslerinin stabilizasyonu ile ilişkilendirilmektedir [10].
2.4 Oksidatif Stresin Azaltılması
Tribenoside, reaktif oksijen türlerinin (ROS) birikimini azaltarak NF-κB fosforilasyonunu baskılar ve DNA hasarını önleyebilir [11].
Bu antioksidan etki, fibroblast, endotelyal ve epitel hücrelerinde gen ekspresyon dengesi sağlayarak anti-inflamatuvar fenotipi destekler.
Dolayısıyla tribenoside, oksidatif stres–inflamasyon döngüsünü kıran bir dengeleyici rol üstlenebilir.
3. Potansiyel Onkolojik Perspektif
3.1 Tümör Mikroçevresi Üzerindeki Olası Etkiler
Tribenoside, doğrudan antineoplastik bir ajan olmamakla birlikte, tümör mikroçevresinde aşağıdaki üç temel eksende etkili olabilir:
• Hipoksi ve Anjiyogenez: Mikrosirkülasyonun düzeltilmesi HIF-1α–VEGF eksenini baskılar, tümör dokusunun oksijenlenmesini artırır.
• Kronik İnflamasyon: Mast hücre stabilizasyonu ve sitokin üretiminin azalması, inflamasyon aracılı tümör büyümesini sınırlayabilir [8,9].
• ECM Stabilizasyonu: Laminin sentezinin artması ve fibroblast aktivasyonunun dengelenmesi, stromal invazyonu fiziksel olarak kısıtlayabilir [10].
3.2 Onkovirüs ve Onkomantar Bağlamı
EBV, HPV ve CMV gibi onkovirüsler, kronik inflamasyon ve vasküler disfonksiyon yoluyla viral persistanlığı sürdürür [12].
Tribenoside’in vasküler bariyer güçlendirici ve inflamasyon baskılayıcı etkileri, bu döngüyü teorik olarak bozabilir.
Benzer şekilde Candida ve Malassezia gibi mantarlar, stromal inflamasyonu ve STAT3 aktivasyonunu artırarak immün baskı oluşturur [13].
Tribenoside, bu mantar kaynaklı inflamatuvar ortamı hafifleterek immün gözetimi yeniden aktive edebilir
4. Tartışma
Tribenoside, doğrudan antitümör bir ilaç olmamakla birlikte, vasküler stabilizasyon, mast hücre dengelemesi, oksidatif stres azaltımı ve ECM yeniden yapılanması gibi etkileriyle tümör mikroçevresini yeniden şekillendiren bir ajan olarak öne çıkmaktadır.
Bu özellikleri, kronik inflamasyonun ve hipoksinin baskın olduğu tümörlerde (ör. EBV pozitif mide kanseri, stromal yoğun meme tümörleri, kolorektal adenokarsinom) potansiyel fayda sunabilir [8,12].
Bununla birlikte, bazı immünmodülatör ajanlarda gözlemlendiği gibi, viral reaktivasyon riski (özellikle CMV) tribenoside tedavilerinde de dikkatle izlenmelidir [14].
Avantajları:
• Mast hücre stabilizasyonu ve sitokin baskılanması
• Endotelyal bariyer güçlendirmesi
• ECM ve bazal membran bütünlüğünün korunması
• ROS ve NF-κB dengesinin düzenlenmesi
Kısıtlılıkları:
• Antineoplastik doğrudan etkinin eksikliği
• Viral reaktivasyon riski
• Klinik kanıt yetersizliği
5. Sonuç
Tribenoside, çok yönlü vasküler ve inflamatuvar düzenleyici etkileri sayesinde yalnızca damar patolojilerinde değil, aynı zamanda tümör mikroçevresinin yeniden şekillendirilmesine yönelik destekleyici stratejilerde değerlendirilebilecek bir moleküldür.
Gelecek araştırmalar, p-ERK, HIF-1α, VEGF, laminin-332, MMP-9 gibi biyobelirteçlerin analiziyle tribenoside’in hipoksik, inflamatuvar ve viral/mantar pozitif tümör modellerinde etkisini test etmelidir.
Bu molekül, immün homeostazı güçlendiren, vasküler stabiliteyi artıran ve rejeneratif süreçleri destekleyen bir “mikroçevre modülatörü” olarak yeni translasyonel araştırmalara adaydır.
Kaynaklar
1. Kestšřanek J. Hemorrhoid management in women: the role of tribenoside + lidocaine. Drugs Context. 2019;8:212602.
2. Lorenc Z, Gökçe Ö. Tribenoside and lidocaine in the local treatment of hemorrhoids. Eur Rev Med Pharmacol Sci. 2016;20:2742–2751.
3. Rabbani G, et al. Pharmacological activity of tribenoside: in vitro and in vivo studies. Arzneimittelforschung. 1976;26:382–386.
4. Procto-Glyvenol Study Group. Effect of tribenoside + lidocaine cream on fibroblast migration and oxidative stress. Eur Rev Med Pharmacol Sci. 2023;27:250–258.
5. Kikkawa Y, Sugiyama Y, Yamada K. Influence of tribenoside on laminin expression in HaCaT cells. Biol Pharm Bull. 2010;33:307–310.
6. Chen W, et al. Role of hypoxia in cancer progression and therapeutic strategies. Front Oncol. 2023;13:1153947.
7. Wang Y, et al. The role of mast cells in the tumor microenvironment. Cancer Lett. 2024;567:216205.
8. Chen Y, et al. Chronic inflammation in cancer: mechanisms and potential therapeutic targets. Nat Rev Clin Oncol. 2022;19(9):545–562.
9. Fang J, et al. Targeting the tumor vasculature: current and future perspectives. Signal Transduct Target Ther. 2022;7:329.
10. Gullo F, et al. Microbiome-fungal interactions in cancer. Cell Mol Life Sci. 2021;78(3):927–940.
11. Rajagopala SV, et al. The human microbiome and cancer. Nat Rev Cancer. 2020;20:745–762.
12. Okuno Y, et al. EBV and cancer: recent advances in pathogenesis and therapeutics. Cancers (Basel). 2023;15(3):987.
13. Pfaller MA, Diekema DJ. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev. 2007;20(1):133–163.
14. Wang Y, et al. CMV reactivation in immunomodulatory therapies. J Infect Dis. 2024;230(4):567–576.

0 YORUMLAR
Bu KONUYA henüz yorum yapılmamış. İlk yorumu sen yaz...