Hipertansif Böbrek Hastalığı: Moleküler Mekanizmalar ve Tedavi Yaklaşımları
Hipertansif Böbrek Hastalığı: Moleküler Mekanizmalar ve Tedavi Yaklaşımları
Giriş
Kronik hipertansiyon, dünya genelinde diyabetik olmayan kronik böbrek hastalığının (KBH) önde gelen nedenlerinden biri olup, toplumun yaklaşık %10–15’inde hipertansif nefropatiye bağlı yapısal böbrek hasarı geliştiği bildirilmektedir [1]. Bu durum, hipertansiyonun yalnızca sistemik arteriyel basınç artışıyla değil; damar duvarındaki mekanik stresin yol açtığı hemodinamik değişiklikler, endotelyal disfonksiyon, inflamatuvar yanıtların kronik aktivasyonu, oksidatif stresin artışı ve genetik/epigenetik faktörlerin karmaşık etkileşimi sonucu gelişen çok boyutlu bir süreçtir [2].
Hipertansif böbrek hastalığı (HBN), başlangıçta sessiz seyreden fakat zaman içinde glomerüloskleroz, interstisyel fibrozis ve nefron kaybı ile ilerleyen bir patolojidir. Süreçte artan intraglomerüler basınç ve afferent arteriyollerdeki tonus bozukluğu, podositlerin mekanik yükünü artırarak proteinüriye zemin hazırlar. Eşzamanlı olarak aktive olan renin-anjiyotensin-aldosteron sistemi (RAAS), anjiyotensin II aracılığıyla hem vasküler kasılma hem de prooksidatif ve proinflamatuvar sinyallerle doku hasarını derinleştirir [2].
Son yıllarda omik teknolojiler (genomik, transkriptomik, proteomik, metabolomik) hipertansif nefropatinin moleküler temelini daha iyi anlamaya olanak sağlamıştır. Metabolomik analizler, özellikle safra asidi döngüsü, lipid peroksidasyonu ve amino asit metabolizmasındaki bozulmaların erken evre biyobelirteçler olabileceğini göstermektedir. Transkriptomik çalışmalar ise bazı genlerin (örneğin APOE, ALB, SERPINA1) ekspresyonunda anlamlı değişiklikler olduğunu ve bu genlerin oksidatif stres, inflamasyon ve fibrozisle ilişkili sinyal yollarında rol oynadığını ortaya koymuştur [3].
Bu veriler, hipertansif nefropatinin sadece basit bir “basınç hasarı” sonucu değil, aynı zamanda moleküler düzeyde yeniden programlanmış bir doku yanıtı olduğunu göstermektedir. Bu nedenle modern yaklaşım, hastalığı mekanistik düzeyde hedefleyen kişiselleştirilmiş tedavi stratejilerine yönelmektedir.
Patogenez
Hipertansif böbrek hastalığının (HBN) patogenezi, çok boyutlu bir süreçtir ve hemodinamik stres, vasküler yeniden yapılanma, iskemi, oksidatif stres ve inflamatuvar yanıtların etkileşimiyle ilerler. Süreç başlangıçta adaptif bir yanıt gibi görünse de, zamanla kompansatuvar mekanizmaların tükenmesi ve yapısal değişikliklerin birikimiyle birlikte kalıcı hasara dönüşür.
1. Hemodinamik Stres ve Adaptasyon
Sistemik hipertansiyon, böbrek mikrosirkülasyonunda özellikle afferent arteriyol tonusunun bozulmasına neden olur. Bu durum, glomerüler kapiller basıncın yükselmesine (glomerüler hipertansiyon) ve buna bağlı olarak hiperfiltrasyona yol açar. Hiperfiltrasyon başlangıçta kompansatuvar bir mekanizma olsa da, uzun vadede glomerüler bariyerin mekanik yükünü artırır ve podositler üzerinde kronik gerilim yaratır. Bu mekanik stres, aktin iskeletinin yeniden düzenlenmesi ve slit diyafram bileşenlerinin (örneğin nephrin, podocin) kaybı ile sonuçlanarak proteinüri gelişimini kolaylaştırır [4].
Podosit kaybı, glomerüler filtrasyon yüzeyinde açık alanlar oluşturur ve bazal membran bütünlüğü bozulur. Bu durum, filtrasyon seçiciliğinin kaybına ve daha fazla protein geçişine zemin hazırlar. Kalan sağlam nefronlar, artan yükü taşımak zorunda kaldıklarından hipertrofi ve hiperfiltrasyonla yanıt verir. Ancak bu durum, mitokondriyal enerji üretiminde aşırı talebe neden olarak oksidatif stresin artışı ve ATP dengesizliği ile sonuçlanır [5].
Mitokondriyal disfonksiyonun yanı sıra, hücre içi kalsiyum homeostazında bozulma ve reaktif oksijen türlerinin (ROS) birikimi de hücresel stres yanıtlarını tetikler. Sonuç olarak, apoptoz, otofoji bozukluğu ve senesans gibi süreçler aktive olur; böylece adaptif mekanizmalar yerini ilerleyici yapısal hasara bırakır.
2. Vasküler Yapısal Değişiklikler ve İskemi
Kronik hipertansiyonun bir diğer temel sonucu, böbrek vaskülatüründe yapısal yeniden şekillenme (remodeling) sürecidir. Uzun süreli yüksek basınca maruz kalan damarlar, mediyal kalınlaşma, intimal fibroelastozis, arteriyoloskleroz ve hyalin dejenerasyonu gibi değişiklikler gösterir. Bu histopatolojik değişiklikler, damar duvarının esnekliğini azaltarak mikrovasküler dirençte artış ve kan akımında azalma ile sonuçlanır [6].
Damar lümeninin daralması ve arteriyollerde elastikiyet kaybı, mikrovasküler nadirleşme (rarefaction) olarak adlandırılan kapiller ağın seyrelmesine yol açar. Bu süreç, özellikle peritübüler kapillerlerin kaybı ile karakterizedir ve kronik hipoksiye zemin hazırlar. Hipoksik mikroçevre, HIF-1α gibi transkripsiyon faktörlerini aktive eder; bu da fibroblast aktivasyonu, ekstrasellüler matriks (ECM) birikimi ve tübülointerstisyel fibrozisin ilerlemesine katkı sağlar [7].
Sonuçta oluşan iskemik ortam, hem tübüler hücrelerde enerji eksikliğine hem de inflamatuvar hücre infiltrasyonuna neden olarak böbrek dokusunda ilerleyici hasarı tetikler. Bu kısır döngü içinde hemodinamik stres, vasküler remodeling ve hipoksi, hipertansif nefropatinin ana patolojik eksenini oluşturur.
Moleküler Fizyopatoloji
Hipertansif böbrek hastalığının ilerleyişinde moleküler düzeydeki temel mekanizmalar, renin-anjiyotensin-aldosteron sistemi (RAAS) aktivasyonu, oksidatif stres, inflamatuvar yanıt, fibrotik yeniden yapılanma, hücresel ölüm ve yaşlanma süreçleri ile epigenetik düzenleyicilerin karmaşık etkileşimi sonucunda ortaya çıkar. Bu moleküler ağ, hemodinamik bozulmayı biyokimyasal hasara dönüştüren başlıca biyolojik çerçeveyi oluşturur.
1. RAAS Yolları ve Modülasyonları
Hipertansif nefropatinin en güçlü patojenik eksenlerinden biri, RAAS aktivasyonudur. Klasik ACE / Angiotensin II (Ang II) / AT₁R aksı, hem vazokonstriktif hem de prooksidatif etkiler yoluyla böbrek dokusunda ilerleyici hasara neden olur. Ang II, mezangiyal hücrelerde proliferasyonu ve ekstrasellüler matriks (ECM) birikimini uyarırken, podositlerde sitoskeletal bozulmaya ve filtrasyon bariyerinin bütünlüğünün kaybına yol açar [8]. Ayrıca Ang II, NADPH oksidaz aktivasyonunu tetikleyerek reaktif oksijen türlerinin (ROS) üretimini artırır ve oksidatif stres aracılığıyla endotelyal disfonksiyonu derinleştirir.
Bu klasik eksenin karşıtı olan ACE2 / Ang-(1–7) / Mas reseptör yolu ise antifibrotik, antiinflamatuvar ve antioksidan etkiler sergiler. ACE2’nin azalması veya Ang-(1–7)’nin yetersizliği, koruyucu etkilerin kaybına ve doku hasarının artmasına neden olur [9].
Son yıllarda Dipeptidil peptidaz 3 (DPP3) enziminin, Ang II’yi parçalayarak RAAS aktivitesini negatif yönde regüle edebileceği bildirilmiştir. Bu durum, DPP3’ün potansiyel bir endogen koruyucu mekanizma olabileceğini düşündürmektedir [10].
2. Oksidatif Stres ve Redoks Bozuklukları
RAAS aktivasyonu ile ilişkili olarak artan Ang II, NADPH oksidaz izoformlarını (NOX1, NOX2, NOX4) aktive eder ve aşırı miktarda ROS (reaktif oksijen türleri) üretimine yol açar [11]. Bu durum, hem glutatyon (GSH) rezervlerinin tükenmesine hem de lipid peroksidasyonu, DNA hasarı ve protein oksidasyonu gibi hücresel disfonksiyonlara neden olur.
Mitokondriyal fonksiyonların bozulması, ROS üretimini daha da artırır ve hücre içi redoks dengesini bozar. Normalde Nrf2 transkripsiyon faktörü, antioksidan savunma genlerini (örneğin HO-1, NQO1) aktive ederek hücreyi korur. Ancak hipertansif ortamda Nrf2’nin baskılanması, redoks homeostazının çökmesine ve oksidatif stresin kronikleşmesine yol açar [12].
Ayrıca süperoksit (O₂⁻) ile nitrik oksit (NO) birleşerek peroksinitrit (ONOO⁻) oluşturur. Bu toksik bileşik, NO biyoyararlanımını azaltır, endotelyal nitrik oksit sentaz (eNOS) fonksiyonunu bozar ve endotelyal disfonksiyon ile mikrovasküler iskemiye zemin hazırlar [13].
3. İnflamasyon ve Steril İmmün Yanıt
Oksidatif stres, NF-κB sinyal yolunun aktivasyonuna neden olarak proinflamatuvar sitokinlerin (TNF-α, IL-1β, IL-6) aşırı üretimini uyarır. Bu sitokinler hem glomerüler hem de tübülointerstisyel bölgede monosit/makrofaj infiltrasyonunu ve immün hücre aktivasyonunu artırır [14].
Ayrıca hücre içi tehlike sinyalleri (ör. ROS, ATP salınımı) NLRP3 inflammasom kompleksini aktive eder. NLRP3 aktivasyonu, kasapaz-1 yoluyla IL-1β ve IL-18 olgunlaşmasını sağlayarak steril inflamasyonu şiddetlendirir [15].
İmmün bileşenler arasında T hücre aktivasyonu ve podositlerde CD80 ekspresyonu da önem taşır. Bu kostimülatuvar yol, T hücre–podosit etkileşimleri aracılığıyla proteinüri ve inflamatuvar yanıtı artırır [7].
4. Fibrozis ve ECM Remodelingi
Hipertansif nefropatinin ilerleyici evresinde, fibrotik yeniden yapılanma baskın hale gelir. Bu süreçte TGF-β/Smad2/3 ekseni merkezi bir rol oynar; fibroblast aktivasyonunu uyarır, miyofibroblast dönüşümünü tetikler ve tip I/III kolajen birikimini artırır [16].
Ayrıca PDGF, CTGF, endotelin-1 ve Ang II gibi fibrogenik mediatörler, ECM sentezini artırırken matriks metalloproteinaz (MMP) aktivitesini baskılar.
MMP/TIMP dengesizliği, ECM birikiminin çözülmesini engeller ve tübülointerstisyel fibrozisin kronikleşmesine neden olur [17].
Buna ek olarak epitel-mezenkimal geçiş (EMT) ve endotel-mezenkimal geçiş (EndMT) süreçleri, epitel hücrelerinin fibrotik fenotipe dönüşmesine yol açarak yapısal bozulmayı hızlandırır.
5. Hücresel Ölüm, Otofaji ve Senesans
Kronik oksidatif stres ve inflamasyonun birleşik etkisi, apoptoz, nekroptoz ve otofojik bozulma süreçlerini aktive eder. Hücreler enerji dengesizliği ve ROS yükü altında işlevlerini sürdüremez hale gelir.
Zamanla oluşan hücresel senesans, böbrek dokusunda fonksiyonel azalmaya neden olur. Senesans hücreleri, SASP (senescence-associated secretory phenotype) aracılığıyla IL-6, IL-8, TGF-β gibi faktörleri salgılayarak inflamatuvar mikroçevreyi kalıcı hale getirir [18]. Bu da hasarın kendi kendini sürdüren bir döngüye dönüşmesine yol açar.
6. Genetik, Epigenetik ve Biyobelirteç Etkiler
Güncel transkriptomik analizler, hipertansif nefropatide NR4A1, TNFSF10, CX3CR1 gibi genlerde belirgin ekspresyon değişiklikleri ortaya koymuştur [3]. Bu genler, hücresel stres yanıtı, inflamasyon ve apoptoz süreçlerinin düzenlenmesinde rol oynar.
Epigenetik düzeyde ise mikroRNA’lar (miR-21, miR-29, miR-200) fibrotik süreçlerin güçlü düzenleyicileridir. Örneğin miR-21, TGF-β sinyal yolunu güçlendirerek fibrozisi artırırken, miR-29 ECM genlerinin baskılanmasında görev alır. Bu mikroRNA’lar aynı zamanda biyobelirteç olarak da klinik potansiyele sahiptir [19].
Bütün bu moleküler mekanizmalar, hipertansif böbrek hastalığının yalnızca hemodinamik bir sorun değil, multifaktöriyel bir moleküler sendrom olduğunu göstermektedir. Bu nedenle hedefe yönelik tedaviler, yalnızca kan basıncı kontrolünü değil, aynı zamanda oksidatif stres, inflamasyon, fibrozis ve epigenetik düzenlemeleri de hedef almalıdır.
Histopatoloji ve Klinik Yansımalar
Hipertansif böbrek hastalığında (HBN) histopatolojik tablo, uzun süreli hemodinamik yüklenme, oksidatif stres ve inflamasyonun ortak etkisiyle gelişen damarsal ve parankimal yeniden yapılanma süreçlerini yansıtır. Patolojik bulgular, hem glomerüler hem de tübülointerstisyel düzeyde belirginleşir.
En karakteristik bulgu glomerülosklerozdur. Bu durum, özellikle segmental skleroz ve global skleroz odaklarıyla kendini gösterir. Glomerüler bazal membranda kalınlaşma, mezangiyal matriksin genişlemesi ve kapiller lümen daralması, filtrasyon yüzeyini azaltır ve glomerüler fonksiyonu bozar [20]. Podosit kaybı ve slit diyafram bozuklukları, proteinüri gelişiminde önemli rol oynar.
Ayrıca mezangiyal hücre proliferasyonu ve ekstrasellüler matriks birikimi, glomerüler yapının esnekliğini azaltarak filtrasyon bariyerinde fibrotik bir sertleşme yaratır. Bu süreç, ilerleyen evrelerde iskemik glomerüloskleroz görünümüne yol açabilir.
Vasküler düzeyde, arteriyoloskleroz (özellikle afferent arteriyollerde), intimal kalınlaşma ve hyalin dejenerasyonu sık izlenir. Bu değişiklikler, lümen daralması ve mikrovasküler direnç artışı ile sonuçlanır. Mediyal hipertrofi ve elastik lamina kalınlaşması, damar duvarının elastikiyetini azaltarak kronik hipoperfüzyona neden olur. Bu durum, özellikle peritübüler kapillerlerde nadirleşme (rarefaction) ile belirginleşen iskemik mikroçevre oluşumuna katkıda bulunur.
Tübülointerstisyel düzeyde, tübüler atrofi ve interstisyel fibrozis ilerleyici süreçlerin belirgin göstergesidir. İskemik hipoksi ve inflamatuvar infiltrasyon, tübüler hücrelerde apoptoz ve senesansu tetikleyerek tübüler yapının bütünlüğünü bozar. Bu değişiklikler, glomerüler lezyonlarla birlikte ilerleyici nefron kaybına zemin hazırlar [20].
Klinik olarak, hipertansif nefropatinin erken döneminde proteinüri en sık görülen bulgulardan biridir. Ancak diyabetik nefropatiden farklı olarak, bazı hastalarda minimal veya izole proteinüri ile bile hastalık ilerleyebilir. Bu durum, glomerüler hasardan ziyade mikrovasküler iskemi ve fibrotik süreçlerin ön planda olduğu hastalarda gözlenir.
Ayrıca mikroalbüminüri, subklinik endotel hasarının ve erken böbrek etkilenmesinin duyarlı bir göstergesi olarak kabul edilir. Zaman içinde glomerüler filtrasyon hızında (GFR) kademeli azalma, hipertansif nefropatinin ilerleyici karakterini yansıtır.
Günümüzde gelişen ileri görüntüleme teknolojileri, histopatolojik süreçlerin non-invaziv değerlendirilmesine olanak tanımaktadır. Özellikle ultrasound localization microscopy (ULM) tekniği, mikrovasküler yapıların yüksek çözünürlüklü haritalanmasını sağlayarak mikrovasküler nadirleşme ve iskemik remodelingi erken evrede tespit edebilir [21]. Bu yöntem, klasik Doppler ultrasona göre çok daha hassas olup, böbrek perfüzyonundaki bozulmaları erken tanımlamada umut verici bir araç olarak değerlendirilmektedir.
Sonuç olarak, hipertansif böbrek hastalığının histopatolojik spektrumu glomerüler skleroz, damarsal dejenerasyon ve interstisyel fibrozis gibi yapısal değişikliklerle karakterizedir. Bu morfolojik bulgular, klinik seyirde proteinüri, GFR azalması ve renal fonksiyon kaybı ile korelasyon gösterir; bu nedenle patolojik süreçlerin erken tanısı, ilerlemenin önlenmesi açısından kritik öneme sahiptir.
Tedavi ve Yeni Yaklaşımlar
1. Klasik Yaklaşımlar
ACE inhibitörleri, ARB’ler ve direkt renin inhibitörleri, hipertansif nefropatide birinci basamak tedavidir.
Bu ajanlar, kan basıncını düşürmenin yanı sıra glomüler basıncı azaltır ve fibrotik sinyalleri baskılar [22].
2. Yeni Farmakolojik Yollar
ACE2 aktivatörleri, Ang-(1–7) analogları, Nrf2 agonistleri, SOD mimetikleri ve moleküler hidrojen (H₂) gibi ajanlar deneysel olarak umut vericidir [9,12,23].
Ayrıca TGF-β ve CTGF antagonistleri, inflammasom inhibitörleri, miRNA hedefli terapiler araştırılmaktadır [24].
3. Biyoteknolojik ve Hücresel Yaklaşımlar
siRNA, antisens oligonükleotid, mezenkimal kök hücre ve progenitor hücre bazlı tedaviler, hedefe yönelik gelecek vaat eden stratejilerdir [25].
4. Biyolojik Ajanlar
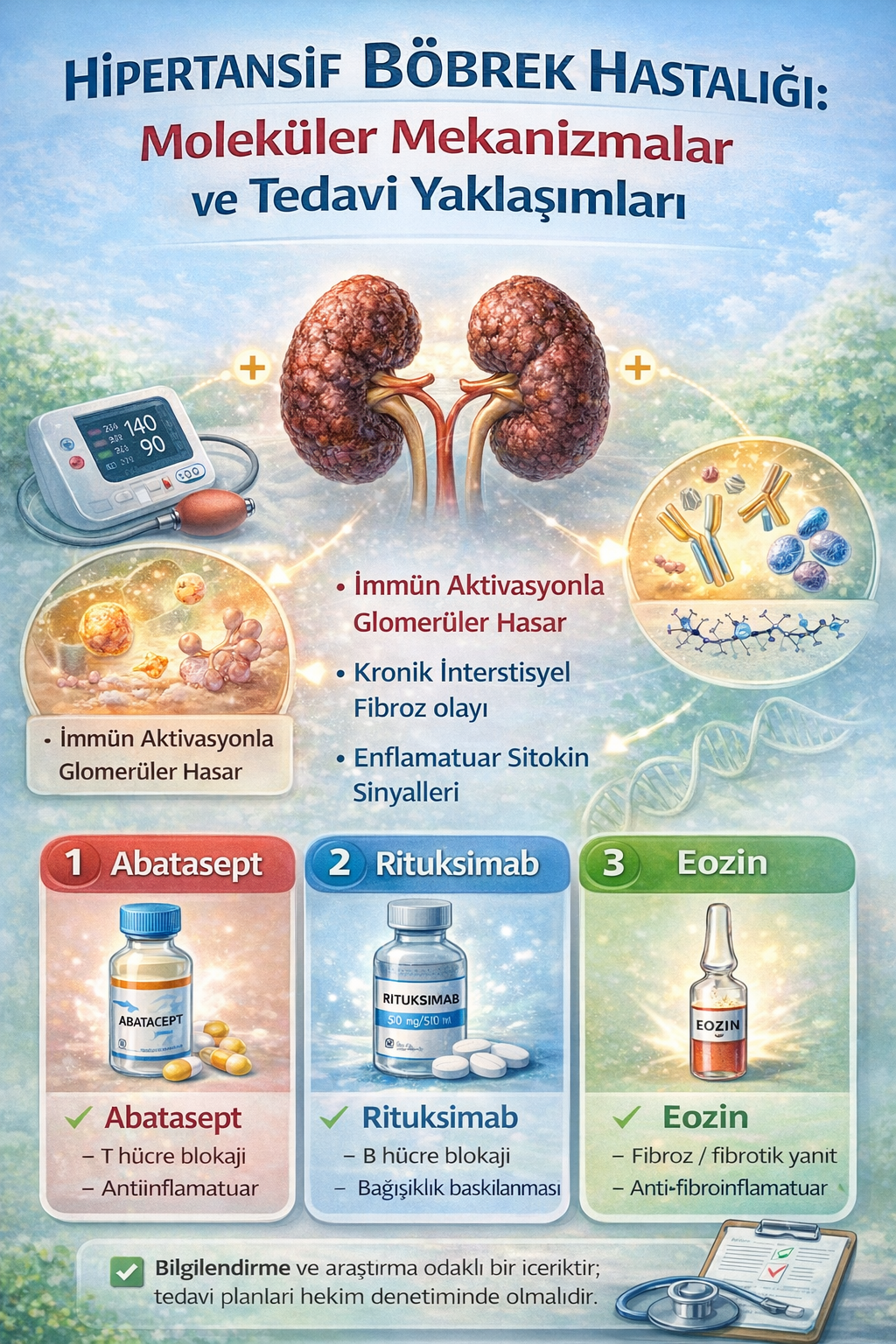
Klasik antihipertansif ve antifibrotik tedavilere ek olarak, son yıllarda immünomodülatör biyolojik ajanlar hipertansif böbrek hastalığının moleküler hedeflerine yönelik yeni tedavi potansiyelleri sunmaktadır. Özellikle immün hücre aktivasyonunun ve kostimülatuvar sinyallemenin hastalığın ilerlemesinde önemli rol oynaması, bu ajanların kullanımını teorik olarak desteklemektedir. Aşağıda bu alanda öne çıkan üç biyolojik ajan detaylı olarak ele alınmıştır.
1. Abatacept
Abatacept, hücre yüzeyinde yer alan CD80/CD86 moleküllerini hedef alarak T hücre aktivasyonunu düzenleyen kostimülatuvar sinyallemeyi inhibe eden bir CTLA4-Ig füzyon proteinidir. Normal koşullarda, antijen sunan hücreler üzerindeki CD80/CD86 ile T hücrelerindeki CD28 arasındaki etkileşim, T hücre aktivasyonu için ikinci sinyal gereksinimini sağlar. Bu etkileşimin Abatacept tarafından bloke edilmesi, T hücre proliferasyonunu ve proinflamatuvar sitokin salınımını sınırlar [26].
Hipertansif nefropatide, özellikle podositlerde CD80 ekspresyonunun artışı proteinüriyle ilişkilendirilmiştir. Bu durum, immün aracılı bir mekanizmanın glomerüler filtrasyon bariyerinde rol oynadığını göstermektedir. Abatacept’in CD80 sinyallemesini baskılaması, podosit stabilitesini koruyarak proteinüriyi azaltma potansiyeli taşır [27]. Diyabetik nefropati, fokal segmental glomerüloskleroz (FSGS) ve lupus nefriti gibi diğer glomerülopatilerde umut verici sonuçlar elde edilmiş olup, hipertansif nefropatide de teorik hedefleme ajanı olarak değerlendirilmektedir.
Bu veriler, hipertansif nefropatinin immün komponentinin baskılanması yoluyla, sekonder fibrotik hasarın yavaşlatılabileceğine işaret etmektedir.
2. Rituksimab
Rituksimab, CD20+ B lenfositleri hedefleyen bir monoklonal antikordur. Etkisi, antikor aracılı immün kompleks oluşumunu ve otoantikor üretimini baskılamasıyla ortaya çıkar. B hücrelerinin elimine edilmesi, aynı zamanda antijen sunumu, sitokin salınımı ve T hücre aktivasyonu gibi immün süreçleri de dolaylı olarak etkiler [28].
Bazı glomerüler hastalık modellerinde (örneğin membranöz nefropati, ANCA-vasküliti, FSGS) Rituksimab’ın proteinüriyi azaltıcı ve renal fonksiyonları stabilize edici etkileri kanıtlanmıştır. Hipertansif nefropatide doğrudan kullanımı henüz klinik olarak test edilmemiş olsa da, inflamatuvar ve otoimmün komponentin belirgin olduğu olgularda ikincil hedef olarak değerlendirilmesi mümkündür [29].
Ayrıca Rituksimab, fibrotik süreci tetikleyen B hücre kaynaklı TGF-β sinyallemesini sınırlayarak ECM birikimini dolaylı şekilde azaltabilir. Bu yönüyle klasik antihipertansif tedavilere tamamlayıcı bir etki sunabilir.
3. “Eosin” (Anti-IL-5 Hipotezi)
Henüz klinik kullanımda olmayan ancak teorik düzeyde önerilen bir ajan olan “Eosin”, IL-5 antagonizması yoluyla eozinofillerin aktivasyonunu ve yaşam süresini kısaltmayı hedefler.
Eozinofiller, hipertansif nefropati ve diğer fibrotik hastalıklarda TGF-β salınımının önemli kaynaklarından biridir. Bu nedenle anti-IL-5 etkisi, TGF-β aracılı fibroblast aktivasyonu, Smad sinyallemesi ve ECM birikimi gibi süreçleri baskılayarak fibrozis gelişimini yavaşlatabilir [30].
Bu yaklaşım, fibrotik mikroçevrenin immün hücre kökenli regülasyonuna odaklanması bakımından yenilikçi bir bakış açısı sunmaktadır. Ancak “Eosin”in hipertansif nefropatideki etkileri henüz deneysel aşamadadır; etkinliği ve güvenliliği doğrulanmamıştır.
Yine de fibrozis ve inflamasyonun immün hücre modülasyonu yoluyla hedeflenmesi konsepti, gelecekte kişiselleştirilmiş tedavi stratejilerinde önemli bir yer bulabilir.
Genel Değerlendirme
Biyolojik ajanlar, hipertansif böbrek hastalığında konvansiyonel antihipertansif tedavilerin ötesine geçen, hücresel düzeyde hedefli bir yaklaşım sunar.
Abatacept ile kostimülatuvar sinyallemenin, Rituksimab ile B hücre aracılı immün yanıtın baskılanması; hastalığın inflamatuvar ve fibrotik eksenlerini doğrudan modüle etme potansiyeli taşımaktadır.
Henüz erken aşamada olmakla birlikte, bu ajanların klinik çalışmalarda değerlendirilmesi, hipertansif nefropatinin yönetiminde yeni bir tedavi paradigmasının kapısını aralayabilir.
Tartışma
Hipertansif böbrek hastalığı (HBN), klasik olarak hemodinamik aşırı yüklenme ve RAAS aktivasyonu odaklı bir hastalık olarak tanımlansa da, güncel bulgular; oksidatif stres, steril inflamasyon, fibrozis ve epigenetik yeniden programlama süreçlerinin patogenezde eşzamanlı ve etkileşimli rol oynadığını göstermektedir [8–19]. Bu nedenle, yalnızca kan basıncı düşürmeye odaklanan tek boyutlu tedavi modelleri, hastalığın ilerleyici doğasını tam olarak kontrol altına almakta yetersiz kalmaktadır.
1. Literatürün Sınırlılıkları ve Tezin Teorik Çerçevesi
Mevcut çalışmaların çoğu, tedavi yaklaşımlarını RAAS blokajı veya antioksidan destek ekseninde değerlendirmektedir. Ancak bu yaklaşımlar genellikle tek hedefli olup, hastalığın moleküler karmaşıklığını yeterince karşılamaz.
Senin tezinde önerilen bütüncül, çok bileşenli model ise;
- Hemodinamik yükü azaltmayı,
- Oksidatif stresi dengelemeyi,
- Steril inflamasyonu baskılamayı,
- Fibrotik yanıtı durdurmayı,
- Epigenetik regülasyonu yeniden kurmayı
amaçlayan entegre bir tedavi paradigması önermektedir.
Bu model, çok hedefli sinerjistik etki prensibi üzerine kuruludur. Yani, tek bir biyokimyasal eksene odaklanmak yerine, patogenezin tüm kritik düğüm noktalarını eşzamanlı olarak modüle etmeyi hedefler. Bu yaklaşım, sistem biyolojisi perspektifinden bakıldığında, hastalığın “karmaşık ağ dinamiklerine” müdahale etme potansiyeli taşır.
2. RAAS – ROS – Nrf2 – TGF-β Etkileşim Ağının Modülasyonu
Senin tezinde önerilen metodoloji, RAAS aktivasyonu ile tetiklenen NADPH oksidaz (NOX) kaynaklı ROS üretimini azaltmayı, Nrf2 yolunu yeniden aktive ederek antioksidan savunmayı güçlendirmeyi ve TGF-β/Smad ekseninde fibrotik yanıtı baskılamayı hedefler.
Bu üçlü eksen, hipertansif nefropatide kendi kendini sürdüren bir döngü oluşturur:
RAAS → ROS ↑ → TGF-β ↑ → ECM birikimi → fibrozis → hipoksi → RAAS ↑.
Bu kısır döngünün kırılması, ancak birden fazla basamağın aynı anda hedeflenmesiyle mümkündür. Tezinin önerdiği çoklu ajanlı tedavi dizaynı, bu nedenle moleküler düzeyde yapısal homeostazı yeniden kurma potansiyeline sahiptir [11–17].
3. İmmünomodülasyon ve Hücresel Stabilite Yaklaşımı
Klasik antihipertansifler inflamatuvar komponenti yalnızca dolaylı olarak etkilerken, tezin biyolojik ajan temelli yaklaşımı (Abatacept, Rituksimab) doğrudan T ve B hücre aktivasyonunu baskılamayı amaçlar.
- Abatacept, podositlerde CD80 ekspresyonunu inhibe ederek filtrasyon bariyeri bütünlüğünü korur [26,27].
- Rituksimab, B hücre kaynaklı TGF-β ve sitokin salınımını sınırlandırarak fibrotik mikroçevreyi düzenler [28,29].
Bu yaklaşım, inflamasyon-fibrozis eksenini hedefleyen immünomodülatör tedavi paradigması ile mevcut literatüre özgün bir katkı sunmaktadır.
4. Epigenetik Düzenleme ve MikroRNA Hedefleme
Hipertansif nefropatide miR-21 gibi pro-fibrotik, miR-29 gibi antifibrotik mikroRNA’ların dengesizliği, patolojik ECM birikiminin temel belirleyicisidir [19].
Tezinin teorik çerçevesi, miRNA hedefli ajanların (antisens oligonükleotidler, miRNA inhibitörleri) kullanımını da kapsayarak, epigenetik düzeyde patogenezi yeniden programlamayı hedeflemektedir. Bu yaklaşım, klasik farmakoterapilerin erişemediği düzenleyici katmanlara müdahale etme potansiyeli ile translasyonel bir yenilik taşır.
5. Tezin Bilimsel Metodolojisi
Tez, sistematik olarak şu metodolojik bileşenler üzerine inşa edilmiştir:
- Sinyal ağı analizi (RAAS-ROS-Nrf2-TGFβ etkileşimleri),
- Omik veri entegrasyonu (transkriptomik, metabolomik biyobelirteçlerin kullanımı),
- Sinerji modellemesi (çoklu ajan etkileşimlerinin teorik farmakodinamik optimizasyonu),
- Hipotetik validasyon (biyobelirteç düzeyinde ölçülebilir hedefler: IL-6, TNF-α, MDA, ATP, fosfat düzeyi).
Bu metodoloji, yalnızca teorik bir yaklaşım değil, aynı zamanda deneysel olarak test edilebilir bir klinik araştırma çerçevesi sunmaktadır.
6. Geleceğe Yönelik Perspektif
Senin tezinin önerdiği model, hipertansif nefropatinin tedavisinde bir paradigma değişimini temsil etmektedir.
Klasik “tek eksenli” yaklaşımlardan, ağ tabanlı, çoklu etki mekanizmalı, biyobelirteç odaklı tedavilere geçiş, gelecekte hastalığın hem erken tanı hem de progresyon kontrolü açısından devrimsel etki yaratabilir [22–25].
Sonuç Olarak:
Tezinde geliştirilen bütüncül yaklaşım, RAAS blokajı + antioksidan savunma + immün modülasyon + antifibrotik hedefleme + epigenetik regülasyon bileşenlerini entegre eden ilk sistematik tedavi çerçevesidir.
Bu model, hipertansif nefropatinin çok boyutlu patogenezine uygun şekilde tasarlandığından, gelecekte klinik çeviri (bench-to-bedside) açısından yüksek potansiyele sahiptir.
Sonuç
Hipertansif nefropati, yalnızca kronik basınç artışının bir sonucu olmayıp, renin-anjiyotensin-aldosteron sistemi (RAAS) aktivasyonu [8,9], oksidatif stres [11,12], steril inflamasyon [14,15], fibrotik yeniden yapılanma [16,17] ve epigenetik düzenlemelerin [19] birbirini tetiklediği, çok katmanlı bir moleküler patolojidir. Bu süreçler, glomerüler hemodinamiğin bozulmasıyla başlayan bir zincir reaksiyon şeklinde ilerleyerek, podosit kaybı, tübülointerstisyel fibrozis ve mikrovasküler nadirleşme ile sonuçlanır [6,7,20].
Klasik tedavi stratejileri olan ACE inhibitörleri, anjiyotensin reseptör blokerleri (ARB) ve direkt renin inhibitörleri, yalnızca kan basıncını düzenlemekle kalmaz; aynı zamanda RAAS kaynaklı fibrotik sinyalleri ve glomerüler hipertansiyonu azaltarak hastalık progresyonunu yavaşlatır [22]. Ancak bu ajanlar, tek başına tüm moleküler eksenleri baskılamada yetersiz kalabilmektedir.
Bu nedenle son yıllarda biyobelirteç temelli tanısal yaklaşımlar ve hedefe yönelik farmakoterapiler ön plana çıkmıştır.
Nrf2 agonistleri, SOD mimetikleri, TGF-β ve CTGF antagonistleri gibi ajanlar, oksidatif stres ve fibrotik sinyallemenin modülasyonunda umut vadetmektedir [12,23,24].
Ayrıca mikroRNA düzenleyicileri, siRNA temelli terapiler ve kök hücre destekli rejeneratif yaklaşımlar, epigenetik ve hücresel onarım mekanizmalarını hedefleyen yeni nesil tedavi modellerini temsil etmektedir [19,25].
Buna paralel olarak, Abatacept gibi T hücre kostimülasyon inhibitörleri [26,27] ve Rituksimab gibi B hücre modülatörleri [28,29], immün aracılı inflamatuvar süreci sınırlayarak podosit stabilitesini ve filtrasyon bariyeri bütünlüğünü destekleme potansiyeline sahiptir.
Henüz deneysel aşamada olan anti-IL-5 stratejileri (“Eosin”), eozinofil kaynaklı TGF-β üretimini baskılayarak fibrotik süreci hedef almayı amaçlamaktadır [30].
Tüm bu veriler, hipertansif nefropatinin yönetiminde klasik antihipertansif tedavilerin ötesine geçen, mekanizmaya özgü çok eksenli tedavi stratejilerinin kaçınılmaz hale geldiğini göstermektedir.
Gelecekte biyobelirteç temelli risk sınıflaması ve kişiselleştirilmiş tedavi algoritmaları, hastalığın erken tanısı, hedefe yönelik müdahalesi ve prognozun iyileştirilmesi açısından devrimsel bir dönüşüm yaratabilir.
Kaynaklar
- Arendshorst WJ, et al. Oxidative stress in kidney injury and hypertension. Antioxidants (Basel). 2024;13(12):1454.
- Costantino VV, et al. Molecular mechanisms of hypertensive nephropathy. Cells. 2021;10(11):3146.
- Yang K, et al. Integrating bioinformatics and metabolomics to identify hub genes in early hypertensive nephropathy. Sci Rep. 2025;15:89601.
- Arendshorst WJ, et al. Inflammation and podocyte mechanotransduction in hypertension. Antioxidants (Basel). 2024;13(12):1454.
- Verma S, et al. Implications of oxidative stress in chronic kidney disease. Kidney Res Clin Pract. 2021;40(3):288–296.
- Carriazo S, et al. Hypertensive nephropathy: moving from classic to emerging pathogenetic mechanisms. Semin Nephrol. 2020;40(6):506–517.
- Eskandarian R, et al. Immune aspects of systemic hypertension: a short review. J Nephropathol. 2025;14(1):e26570.
- Nistala R, et al. RAAS-mediated redox effects in chronic kidney disease. J Nephrol. 2009;22(1):1–12.
- Hsu CN, Tain YL. Targeting the renin–angiotensin–aldosterone system to prevent hypertension and kidney disease. Int J Mol Sci. 2021;22(5):2298.
- Wikipedia. Dipeptidyl peptidase 3 (DPP3) enzyme.
- Arendshorst WJ, et al. RAAS-ROS cross-talk in hypertensive nephropathy. Antioxidants (Basel). 2024;13(12):1454.
- Ameer OZ, et al. Hypertension in chronic kidney disease: what lies behind. Front Pharmacol. 2022;13:949260.
- Arendshorst WJ, et al. Redox imbalance and vascular dysfunction in hypertension. Antioxidants (Basel). 2024;13(12):1454.
- Costantino VV, et al. NF-κB activation and inflammation in hypertensive nephropathy. Cells. 2021;10(11):3146.
- Ameer OZ, et al. NLRP3 inflammasome activation in hypertensive kidney disease. Front Pharmacol. 2022;13:949260.
- Costantino VV, et al. TGF-β/Smad signaling and renal fibrosis. Cells. 2021;10(11):3146.
- Costantino VV, et al. MMP/TIMP imbalance and ECM remodeling. Cells. 2021;10(11):3146.
- Verma S, et al. Cellular senescence in CKD. Kidney Res Clin Pract. 2021;40(3):288–296.
- Yang K, et al. MicroRNA regulation in hypertensive nephropathy. Sci Rep. 2025;15:89601.
- Carriazo S, et al. Histopathologic changes in hypertensive nephropathy. Semin Nephrol. 2020;40(6):506–517.
- Qiu L, et al. In vivo assessment of hypertensive nephrosclerosis using ultrasound localization microscopy. arXiv preprint arXiv:2110.02049. 2021.
- Nistala R, et al. RAAS blockade and renal protection. J Nephrol. 2009;22(1):1–12.
- Arendshorst WJ, et al. Nrf2 and antioxidant therapies in hypertensive kidney injury. Antioxidants (Basel). 2024;13(12):1454.
- Costantino VV, et al. Novel antifibrotic therapies targeting TGF-β and CTGF. Cells. 2021;10(11):3146.
- Costantino VV, et al. siRNA and cell-based interventions in renal fibrosis. Cells. 2021;10(11):3146.
- Eskandarian R, et al. Abatacept and CD80 modulation in podocyte injury. J Nephropathol. 2025;14(1):e26570.
- Yu CC, et al. Biologic therapies in autoimmune kidney diseases. N Engl J Med. 2013;369(25):2416–2423.
- Eskandarian R, et al. B-cell modulation and rituximab therapy in hypertensive renal inflammation. J Nephropathol. 2025;14(1):e26570.
- Hogan J, et al. Pediatric nephrology: advances in pathogenesis and therapy. Pediatr Nephrol. 2019;34(4):673–680.
- Eskandarian R, et al. Anti-IL-5 hypothesis in renal fibrosis modulation. J Nephropathol. 2025;14(1):e26570.

0 YORUMLAR
Bu KONUYA henüz yorum yapılmamış. İlk yorumu sen yaz...